用于scATAC-seq有监督分类的Cellcano
细胞类型识别是单细胞数据分析的基本步骤。由于高质量参考数据集的可用性,有监督细胞分类方法在scRNA-seq数据中很受欢迎。染色质可及性分析(scATAC-seq)的最新技术进步为理解表观遗传异质性带来了新的见解。随着scATAC-seq数据集的不断积累,迫切需要专门为scATAC-seq设计的有监督细胞分类方法。作者开发了Cellcano,这是一种基于两轮监督学习的算法,用于从scATACseq数据中识别细胞类型。
来自:Cellcano: supervised cell type identification for single cell ATAC-seq data
表观遗传异质性:细胞或个体内同一基因组中某些基因的表达水平因表观遗传机制(如DNA甲基化、组蛋白修饰等)而出现的差异。这些表观遗传差异可以影响基因的转录和翻译,从而导致表型的差异,包括细胞命运、发育、生长、代谢和疾病等方面的变化。
scRNA-seq也具有表观遗传异质性,scATAC可以作为该异质性的另一种体现。
目录
- 背景概述
- 方法
- Cellcano架构
- ATAC细胞分型任务与基因特征空间
- ATAC的另外两种特征空间
背景概述
单细胞测序技术的发展极大地增强了对复杂组织生物学机制的理解。在所有单细胞分析中,scRNA-seq最受欢迎,目前已经开发了1200多种分析工具。在scRNA-seq分析中,基于单个细胞的基因表达值做细胞类型判别(以下称为"细胞分型")是最基本的问题。目前有许多细胞分型方法可用。这些方法可以分为有监督和无监督两类。部分基准研究表明,有监督的细胞分型方法在准确性、鲁棒性和可扩展性方面比无监督的方法具有优势。
基因表达可以由几个因素调节。其中,染色质可及性对于DNA和调控元件之间的相互作用至关重要,并为理解转录调控机制提供了重要信息。近年来,通过scATAC-seq分析,也见证了从测量批量样品中的染色质可及性到单细胞分辨率的转变。与scRNA-seq一样,细胞分型也是scATAC-seq数据分析中的一个重要问题。然而,scATAC-seq数据具有某些特性,这使得细胞分型更加困难。首先,由于低读取计数,scATAC-seq数据要稀疏得多,这导致用于区分细胞类型的信号较弱。其次,与scRNA-seq不同,scATAC-seq数据中的特征空间定义不明确,这给提取有用信息带来了困难。原始scATAC-seq数据可以总结为全基因组固定大小bin的计数、peak代表可访问区域或基因。因此,特征空间的确定是scATAC-seq细胞分型的另一个重要步骤。尽管可以利用多组学测序技术的信息进行细胞分型,但这些数据集昂贵且有限。因此,迫切需要专门为scATAC-seq细胞分型开发方法。
大多数scATAC-seq细胞分型方法都是无监督的,并且基于先验知识。到目前为止,虽然已经开发了许多用于单细胞多组学数据整合的方法,而专门用于scATAC-seq细胞分型的方法非常有限。Seurat和scJoint使用scRNA-seq作为参考,将 label 在联合embedding空间中转移到scATAC-seq。由于不同测序技术之间数据分布的强烈变化,这两种方法可能表现不佳。Signac是最近开发的端到端scATAC-seq数据分析管道,提供scATAC-seq批次整合和标签转移功能。EpiAnno最近也提供了以scATAC-seq为参考在scATAC-seq中进行有监督学习的细胞分型。Signac和EpiAnno的一个主要问题是,它们使用来自所谓peak的读取计数作为输入,其中峰值高度依赖于数据。由于技术和生物伪影,reference和query之间的峰值一致性可能较低,这将导致信息丢失和不理想的细胞分型结果。此外,EpiAnno对于大型数据集在计算上是不可用的。
因此,作者开发了一种用于scATAC-seq的细胞分型方法,名为Cellcano。Cellcano实现了两轮监督学习算法。首先在参考数据集上训练多层感知器,并预测目标数据中的细胞类型。从预测结果中看,Cellcano选择一些被认为预测良好的目标细胞(称为anchor)来形成新的训练集。接下来,Cellcano使用预测的伪标签在anchor上训练自知识蒸馏模型(KD模型),然后应用训练的KD模型来预测剩余的非anchor细胞的细胞类型。通过广泛的真实数据分析,证明了与现有方法相比,Cellcano明显更准确、计算效率更高、可扩展性更强。
方法
Cellcano架构
Cellcano使用来自原始scATAC-seq数据的gene-level summaries作为输入。在给定原始数据的情况下,Cellcano结合了ArchR管道来处理原始数据,并获得参考数据集和目标数据集的gene score。使用gene score可以提供良好的预测准确性和计算效率。然后,Cellcano对参考gene score应用 F-test,选择细胞类型特异性基因作为模型构建的特征。在获得所选特征的参考基因和目标基因得分后,Cellcano采用两轮监督细胞分型策略,如图1所示。在第一轮中,Cellcano用参考基因得分训练MLP模型,并预测目标数据中的细胞类型。如果目标规模太小,Cellcano会停止并返回预测结果。当目标规模足够大(例如,超过1000个细胞)时,Cellcano会执行另一轮模型训练以提高预测结果。第二轮从选择anchor cell开始。为此,首先根据第一轮预测的预测概率计算每个细胞的熵,然后选择熵较低的细胞作为anchor(假设具有较低预测熵的细胞更有可能被准确预测)。然后,使用anchor及其预测的细胞类型作为新的参考数据来训练另一个分类器来预测非anchor细胞。在这里,使用KD模型作为分类器,因为当参考数据具有不完美的标签时,它可以更好地工作。第二轮中的假设是,与在参考数据集上训练的分类器相比,在anchor(来自target数据)上训练的分类器可以更好地捕捉目标数据集中的数据分布,从而提高预测性能。
在F检验中,通过计算两个或多个组的方差比值,来检验样本组之间是否存在显著差异。如果F检验的p值小于预设的显著性水平(通常为0.05或0.01),则可以拒绝零假设,认为不同组之间的差异是显著的,反之,则认为差异不显著。
F检验广泛应用于多个组之间的差异分析,例如,在生物医学研究中,可以用F检验来比较不同组之间的基因表达水平、蛋白质表达水平。
ATAC细胞分型任务与基因特征空间
作者收集并处理了四个人类外周血单核细胞(PBMC)数据集和两个小鼠大脑数据集。对于这六个数据集,设计了50个细胞分型任务,全面覆盖了不同的真实应用场景。
scATAC-seq数据可以在三个不同的特征空间中表示:全基因组固定大小仓(genome-wide fixed-size bins)、峰值(peaks)和基因(genes)。全基因组固定大小的仓具有非常大的特征空间,这造成了沉重的计算负担。峰值不是预先定义的,并且需要调用统一峰值的额外步骤。更重要的是,由于每个数据集的峰值都不同,因此不能对新的目标数据重用预先训练的预测模型。在这项工作中,作者选择基因得分作为输入。此外,还可以将基于基因评分训练的模型进一步连接到scRNA-seq模型。总结基因评分有不同的方法,第一个问题是如何利用这些基因评分模型。ArchR提供了54种基因评分模型的变体,其推荐的模型被证明是在匹配的scATAC-seq和scRNA-seq数据中推断基因表达最准确的。从实际数据分析中,发现使用ArchR推荐的基因评分模型可以从Cellcano获得良好的细胞分型性能。
在scATAC-seq数据中,每个细胞的开放染色质区域可以看作是该细胞特定的基因调控区域。因此,可以通过这些开放区域对单个细胞的基因调控水平进行建模,并得到基因评分。具体来说,基因评分模型通常包括以下几个步骤:
- 建立基因-开放区域映射:首先,需要将每个开放区域与其可能调控的基因进行映射。这可以通过将开放区域与最近的基因或者包含该开放区域的基因进行关联来实现。
- 计算基因调控得分:然后,对于每个基因,可以将与该基因调控相关的开放区域的开放程度进行加权平均,得到基因调控得分。这个得分可以反映该基因在单个细胞中的表达水平。
基因评分的获取可以理解为将ATAC模态转移到RNA模态。
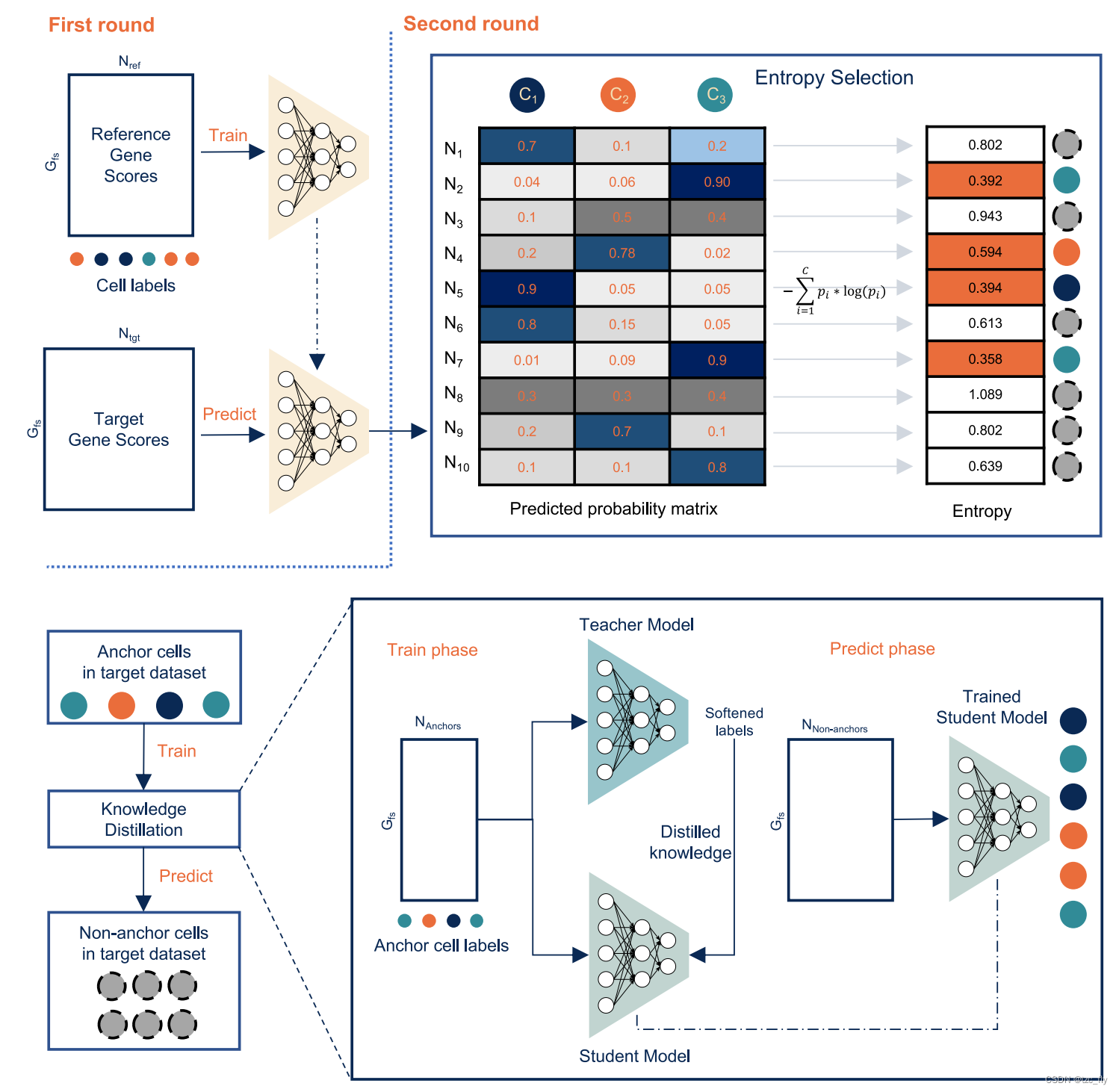
- 图1:Cellcano采用两轮学习策略。在第一轮中,Cellcano根据已知细胞标记的参考基因评分训练MLP。然后,Cellcano使用经过训练的MLP来预测目标基因评分上的细胞类型。当目标数据规模足够大时,Cellcano通过选择锚来开始第二轮学习。利用从第一轮预测获得的预测概率矩阵,计算每个细胞的熵。选择熵相对较低的细胞作为锚,以训练知识蒸馏(KD)模型。训练后的KD模型用于预测剩余非锚中的细胞类型。
ATAC的另外两种特征空间
对于scRNA-seq的表达矩阵,特征是基因,但scATAC-seq的表达矩阵特征定义不止是基因(或者上面说的基因评分),还有peaks,和genome-wide fixed-size bins。
首先介绍peaks空间,在scATAC-seq数据中,表达矩阵的特征是基因组上的开放峰(open chromatin peaks)。开放峰是指基因组上具有染色质开放性的DNA区域,通常与基因的调控相关。在scATAC-seq数据中,对于每个细胞,我们可以通过比较其基因组上的开放峰来获得细胞类型和状态的信息。因此,scATAC-seq数据中的表达矩阵特征可以被看作是基因组上开放峰的集合,而不是RNA模态中基因的集合。每个细胞的开放峰矩阵的值表示该细胞中该开放峰的可及性或开放程度。与scRNA-seq类似,scATAC-seq数据的分析也可以使用一些单细胞分析工具,如Seurat等。
另外考虑genome-wide fixed-size bins空间,ATAC表达矩阵中的genome-wide fixed-size bins是指对基因组进行固定大小的分割(分箱,bin),将每个bin作为特征进行分析。在分析过程中,ATAC-seq数据中的每个细胞都会被分配到一系列bin上,而每个bin则代表了基因组的一个小片段。在每个bin上可以计算每个细胞中reads的数量,从而将每个细胞映射到一个基因组宽度固定的特征向量中。因此,genome-wide fixed-size bins作为特征的含义是将基因组分割为固定大小的区域,并将每个区域中的ATAC-seq信号作为特征,用于单细胞ATAC-seq数据的分析。
在ATAC-seq数据中,每个bin上细胞中reads的数量可以表示为该细胞中在对应基因组位置的染色质开放度。开放的染色质区域通常与活跃的基因表达有关,因此这些数量可以用于研究细胞类型和状态之间的差异。同时,每个bin上reads的数量也可以用于计算peaks;
peaks空间可以看作是size不固定的genome-wide fixed-size bins空间,它们的特征都是表示染色质开放程度。