Nature Communications 单细胞算法 scDist,教你怎么找到重要的细胞亚群与基因!
生信碱移
scDist: 寻找关键细胞亚群与基因的方法
单细胞RNA测序(scRNA-seq)使我们能够研究受药物治疗、感染以及癌症等疾病中关键的细胞亚群。为了找到可能影响疾病的细胞亚群乃至基因,我们常常去比较两个或多个组之间显著差异的细胞类型。这里"显著差异"的定义可以是不同方面的,总结成两种:
-
① 细胞比例差异,如 DA-seq、Milo和Meld,侧重于识别在不同条件下比例变化的细胞类型;
-
② 细胞状态差异,与细胞比例相反,旨在检测在不同条件下具有不同转录组特征的细胞类型。常见的做法是使用不同条件下差异表达基因的数量来评估某种细胞亚群状态的转录状态变化,当然也有一些相关算法被开发出来比如 Augur。
直觉上看,基于转录表达状态的细胞状态方法似乎更合理。之前比较有名的算法Augur,通过训练一个分类器,从表达数据中预测条件标签,然后使用ROC曲线下面积作为指标对细胞类型进行排名。尽管如此,Augur并未考虑个体间的变异性,这可能会混淆扰动细胞类型的排名。来自美国新墨西哥大学的研究者使用六个健康对照者的血液scRNA-seq数据演示了这个现象。通过将六个健康对照者的样本随机分为两组,生成一个假的对照数据集,逻辑上讲算法不应该检测到任何细胞类型的差异。但他们的实验结果表明,在20次随机重复中,93%的细胞类型AUC均大于0.5,红细胞(RBCs)在所有20次试验中均被识别有意义的细胞。
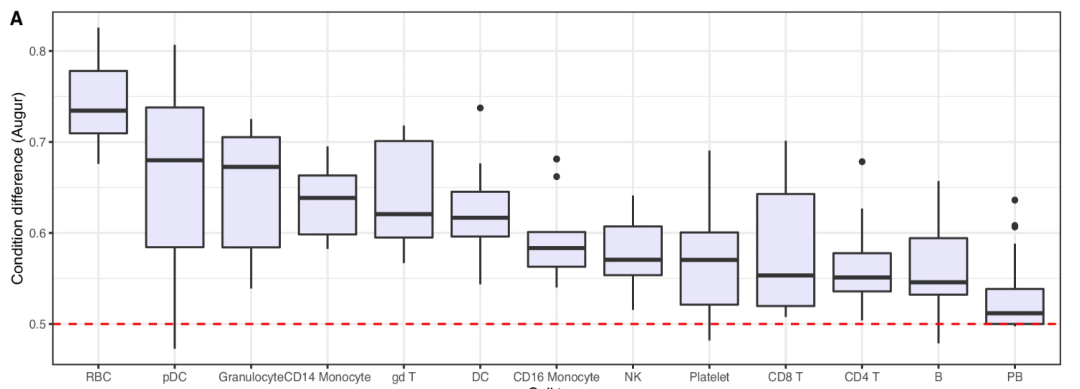
▲ 模拟实验结果:理论上讲每种细胞AUC应该都在0.5附近,因为分组是假的。但是实际上,RBC的平均AUC甚至可以超过0.7.

▲ DOI:10.1038/s41467-024-51649-3
为此,他们开发了一种基于混合效应模型算法scDist,于今年9月1号发表于Nature Communications[IF:14.7]杂志。简单来讲,scDist估计每种细胞类型在高维基因表达空间(如PCA降维空间)中条件均值之间的距离,并采用线性混合效应模型来考虑不同样本和其他技术潜在的变异性。除了找到不同条件下变化最大的细胞亚群,作者还根据主成分回归的beta值与基因在主成分的载荷乘积定义了特定细胞中的基因重要性(基因-->主成分-->条件差异)。
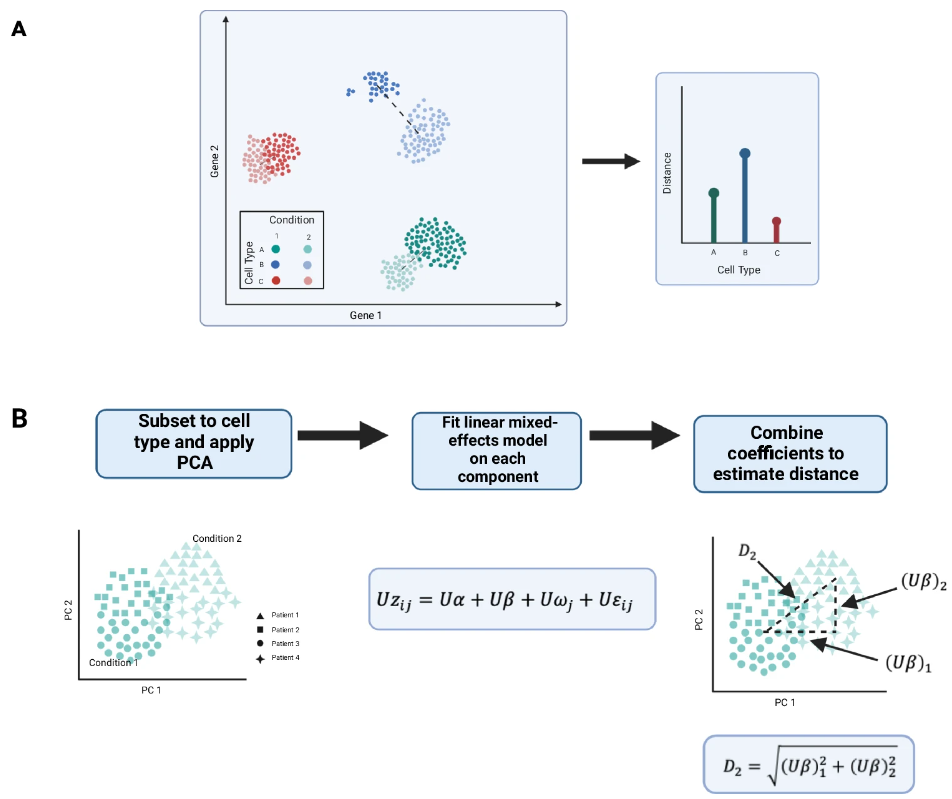
▲ scDist的原理。
小编将在下文简单介绍一下scDist的使用,感兴趣的铁子可以阅读原文或者在下方链接进一步查看:
-
https://github.com/phillipnicol/scDist
0.R包的安装与引用
可以使用下方注释代码安装该包,当然本文还使用了其它包(自行安装):
#devtools::install_github("phillipnicol/scDist")
library(dplyr)
library(Seurat) # v4版本
library(scDist)
library(ggplot2)
library(ggpubr)
1.数据的加载
先读取以下数据,走一下标准的seurat流程。需要注意的是,尽管该方法scDist与任何归一化方法兼容,但作者建议使用scTransform进行归一化。
af <- readRDS("af.rds")
af <- SCTransform(af, vars.to.regress = "percent.mt", verbose = FALSE)
af <- RunPCA(af, verbose = FALSE)
af <- RunUMAP(af, dims = 1:30, verbose = FALSE)
af <- FindNeighbors(af, dims = 1:30, verbose = FALSE)
af <- FindClusters(af, verbose = FALSE, resolution = 0.05)
DimPlot(af, label = TRUE)
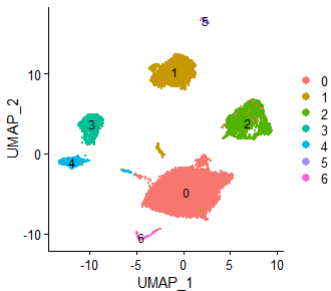
▲ 示例数据的初步分群。
2.执行scDist分析
① 首先从seurat对象af中提取数据构建一个列表sim:
sim <- list(Y=af@assays$SCT@scale.data %>% as.data.frame(),
meta.data=af@meta.data %>% as.data.frame())
# 看一下数据的长相
dim(sim$Y) #Normalized counts
#[1] 3000 18497
head(sim$meta.data)
# orig.ident nCount_RNA nFeature_RNA Type percent.mt percent.rb nCount_SCT nFeature_SCT SCT_snn_res.0.05 seurat_clusters
#AAACATCGAACGCTTACGAACTTA_1 cABMR 1965 789 cABMR 2.340967 1.1195929 1328 757 1 1
#AAACATCGAATCCGTCCCTAATCC_1 cABMR 4828 1558 cABMR 8.243579 5.1367026 1334 681 4 4
#AAACATCGAATGTTGCGATGAATC_1 cABMR 1029 489 cABMR 1.068999 1.1661808 1028 489 1 1
#AAACATCGACACGACCCCGACAAC_1 cABMR 1448 680 cABMR 1.933702 2.4861878 1237 680 1 1
#AAACATCGACAGCAGATGGTGGTA_1 cABMR 7451 2137 cABMR 9.126292 13.7699638 1273 575 4 4
#AAACATCGACCACTGTGTCTGTCA_1 cABMR 1059 457 cABMR 3.493862 0.7554297 1049 457 1 1
② 执行scDist分析,需要根据自己数据设置的参数有d, fixed.effects, random.effects, clusters(仔细看我的注释):
out <- scDist(normalized_counts = sim$Y, # 标准化的数据矩阵
meta.data = sim$meta.data, # metadata表格
d = 20, # 指定用于PCA分析的维度数量为前20
fixed.effects = "orig.ident", # 你感兴趣的分组条件,对应metadata表格中的列名称
random.effects = c(), # 需要去除的潜在的影响因素,比如不同的样本、年龄、批次等等,对应metadata表格中的列名称
clusters="seurat_clusters" # 待分析的细胞类型,对应metadata表格中的列名称
)
③ 可视化分析结果,首先是看看不同细胞类型的在分组间的距离得分:
p <- DistPlot(out, return.plot = TRUE)
p + theme_bw()
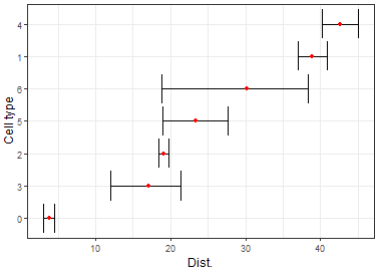
▲ 细胞类型的分组距离得分。
随后可以评估一下基因的重要性,通过out$vals对象中的载荷以及beta值相乘计算。这个计算过程包装在distGenes函数,需要使用cluster参数指定要分析的细胞:
distGenes(out, cluster = "4") # 指定4这一群细胞
df <- data.frame(value = out$vals[[cluster]]$beta.hat,
label = out$gene.names) %>% top_n(20, abs(value)) # 可视化前20个
df$color <- ifelse(df$value>0, "Positive", "Negative")
ggbarplot(df,
x="label",
y="value",
fill = "color",
color = "white",
palette = "npg",
sort.val = "asc",
sort.by.groups = FALSE,
xlab = "",
legend.title = "")+ theme_bw() + ylab("Condition difference") + coord_flip()
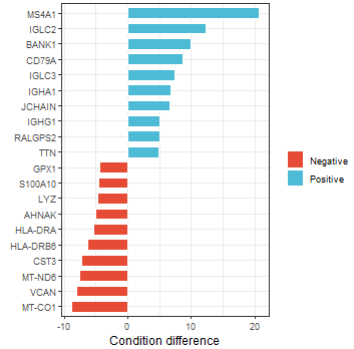
▲ 指定亚群4中的基因重要性得分。
简单分享到这各位老哥老姐
欢迎关注