顶刊精析|METI:整合细胞形态与空间转录组学的肿瘤微环境分析框架·24-09-06
小罗碎碎念
本期精读文献:《METI: Deep profiling of tumor ecosystems by integrating cell morphology and spatial transcriptomics》
今天分享的这篇文献于2023年8月25日发表在Nat Commun,目前IF=14.7。

| 作者类型 | 作者姓名 | 单位名称(中文) |
|---|---|---|
| 第一作者 | Jiahui Jiang | 德克萨斯大学MD安德森癌症中心遗传医学系,美国休斯顿 |
| 通讯作者 | Linghua Wang | 德克萨斯大学MD安德森癌症中心遗传医学系,美国休斯顿 |
| 通讯作者 | Jian Hu | 埃默里医学院人类遗传学系,美国亚特兰大 |
文献速览
这篇论文介绍了Morphology-Enhanced Spatial Transcriptome Analysis Integrator (METI),这是一个端到端的框架,通过整合细胞形态学和空间转录组学,更深入地分析肿瘤微环境中的细胞类型和状态。
研究背景
- 问题:空间转录组学(ST)技术提供了肿瘤微环境(TME)内细胞相互作用的宝贵见解,但大多数现有分析工具缺乏组织学特征的考虑,依赖于匹配的单细胞RNA测序数据,限制了它们在TME研究中的有效性。
- 难点:现有方法通常无法同时利用基因表达数据和组织形态学信息,导致在细胞类型识别和状态分析中存在局限性,特别是在处理低质量数据时。
- 相关工作:现有的空间转录组学平台包括基于下一代测序(NGS)的方法(如Visium、GeoMx、Slide-Seq)和基于杂交的方法(如MERFISH、seqFISH、CosMx)。尽管这些方法在各自领域取得了显著进展,但它们在细胞类型识别的准确性和数据整合方面仍存在不足。
方法
- METI框架结合了空间转录组学、细胞形态学和预定义的基因签名,系统分析癌细胞和TME成分,分层细胞类型和状态,并分析细胞共定位。
- 输入包括基因表达数据、H&E图像和每个斑点的XY坐标。
- METI通过五个模块逐步分析TME:
- 模块1:识别正常和癌前细胞,如胃中的杯状细胞。通过结合形态学特征和基因表达数据进行综合分析,克服了单一数据源的局限性。
- 模块2:识别肿瘤细胞富集区域并表征其异质性。利用特定的肿瘤标记基因(如细胞角蛋白、EPCAM、三叶因子)来识别和表征肿瘤细胞。
- 模块3:空间映射T细胞,包括CD4+和CD8+ T细胞及其状态(如调节性T细胞和耗竭T细胞)。通过特定的T细胞标记基因和免疫检查点基因来识别和表征不同状态的T细胞。
- 模块4:深入分析其他免疫细胞,如中性粒细胞、B细胞、浆细胞和巨噬细胞。利用已验证的基因签名来识别特定免疫细胞类型/状态。
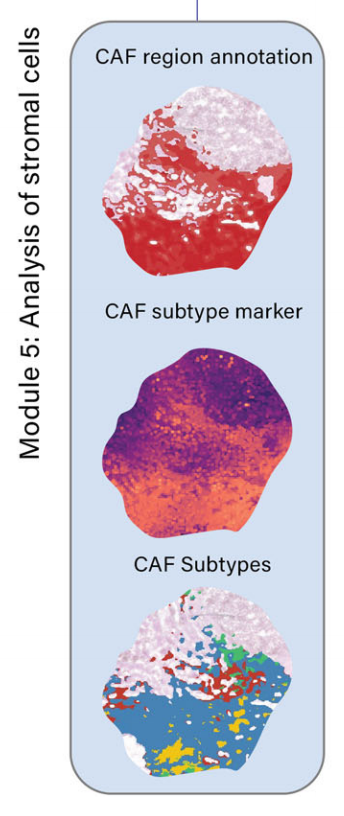
- 模块5:分析癌症相关成纤维细胞(CAF)及其亚型(如myCAFs、iCAFs、apCAFs)。通过特定的成纤维细胞标记基因来识别和表征不同亚型的CAF。
实验
- METI在多种肿瘤组织生成的ST数据上进行了性能评估,包括胃癌、肺癌和膀胱癌以及癌前组织。
- 通过与现有的聚类和细胞解卷积工具进行定量比较,展示了METI的稳健性和一致性。例如,在胃癌样本G1中,METI成功识别了所有四个富含杯状细胞的区域,而基于基因表达数据的TESLA方法未能识别出区域4中的杯状细胞。
- 在肺癌样本L1中,METI识别并可视化了CD4+ Tregs和CD8+ Tex细胞的空间分布,显示出不同样本中T细胞状态的变异性。
结果与分析
- METI在细胞类型识别方面表现出更高的准确性,并且在某一模态数据质量较低时仍保持稳健。例如,在胃癌样本G2中,METI的总体准确率为0.778,而BayesSpace和SpaGCN的最高准确率分别为0.704和0.734。
- METI能够通过整合基因表达和图像分析结果,克服低UMI计数带来的局限性,提供更准确的细胞类型识别。例如,在膀胱癌样本B1和B2中,METI成功识别并注释了中性粒细胞富集区域,与病理学家的标注高度一致。
代码&数据
代码链接
- GitHub仓库:
- 链接:https://github.com/Flashiness/METI
- 作用:包含METI框架的所有原始代码,供研究人员下载和使用。
- Zenodo DOI:https://doi.org/10.5281/zenodo.11247565
数据集
-
肺癌数据集 (LC_1):
- 链接:https://ega-archive.org/studies/EGAS00001005021
- 作用:用于评估METI在肺癌组织中的性能。
- 描述:该数据集来自MD安德森癌症中心,包含了肺癌和正常肺组织的样本。
-
胃癌和膀胱癌数据集:
- 链接:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE246011
- 作用:用于评估METI在胃癌和膀胱癌组织中的性能。
- 描述:这些数据集包含了原始测序和成像数据,已上传至GEO数据库。
一、绪论
空间转录组学(ST)在保留空间信息的同时测量基因表达,这是传统单细胞RNA测序(scRNA-seq)所不具备的1。
空间位置有助于揭示肿瘤微环境(TME)的细胞组成与组织结构,便于探究不同肿瘤区域的空间基因表达模式和细胞相互作用2–4。
常用的ST平台大致可分为两类:基于下一代测序(NGS)的方法,如Visium5、GeoMx6、Slide-Seq7,以及基于原位杂交的方法,如MERFISH8、seqFISH9、CosMx10。NGS-based ST方法虽覆盖整个转录组,但分辨率不足以达到单细胞水平;而原位杂交方法虽具有更高的空间分辨率,但仅限于基因组的一小部分,限制了其在探索性研究中的潜力。
许多ST平台允许对同一组织切片的苏木精-伊红(H&E)染色图像进行高分辨率扫描,这对下游分析极为宝贵。通过仔细检查H&E图像,可以识别出不同细胞类型,无需进行基于基因表达的细胞去卷积分析。
此外,将组织学特征纳入ST数据分析,可以更有效地解决潜在的技术和分析伪迹问题。
为整合空间基因表达与图像,已开发出多种先进方法。
- MUSE11通过深度学习方法分析ST数据中的形态学与转录状态,以表征组织成分;
- BayesSpace12采用完全贝叶斯统计方法,利用空间邻域信息提高ST数据的分辨率并进行聚类分析;
- SpaGCN13利用图卷积网络(GCN)整合基因表达与组织学信息,识别空间域;
- TESLA14采用卷积神经网络整合基因表达与组织学信息,在图像像素级别上绘制肿瘤核心、边缘及不同细胞类型;
- Robust Cell Type Decomposition(RCTD)15利用scRNA-seq数据导出的细胞类型轮廓分解细胞类型,并调整测序技术间的变异;
- CytoSPACE16旨在将scRNA-seq图谱中的单个细胞与空间表达谱对齐。
尽管这些方法表现卓越,但存在若干局限性。
首先,它们是通用分析工具,适用于任何组织类型的数据,而非专门针对癌细胞和TME的研究。未能融入癌症基因组学的领域知识,可能导致这些方法忽略特定于癌细胞和TME中其他关键组分的特征。
例如,scRNA-seq可能无法捕获组织环境中的某些细胞类型和状态,且重要细胞类型可能无法与聚类算法识别的任何簇对齐。此外,某些方法依赖于scRNA-seq数据和细胞注释的可用性,这限制了这些方法在不同研究背景下的灵活性和实用性。上述限制束缚了作者团队对于TME中关键组分和复杂相互作用的表征。
在本研究中,作者团队提出了一种分析框架,通过整合空间基因表达、组织病理学和癌症及TME细胞的先验知识,系统地分析癌细胞和TME细胞。作者团队的方法首先识别TME内的关键细胞组分及其状态,包括各种免疫细胞及其转录状态、肿瘤间质组分如癌症相关成纤维细胞(CAFs)和上皮细胞组分。
形态学增强的空间转录组分析整合器(METI)还提供了H&E图像中各种细胞类型的细胞形态学补充信息。基因表达与组织学特征的结合结果,为组织内空间细胞组成与组织提供了全面的理解。METI在由胃癌、肺癌和膀胱癌等多种癌症类型生成的ST数据集上的评估显示,其性能稳健且一致(补充表1)。
二、结果
METI的工作流程以系统化、逐步的方式分析肿瘤微环境(TME),重点关注从正常细胞到前恶性细胞,再到恶性细胞的进展过程,并检查每个组织切片中的淋巴细胞。
METI以标准ST数据为输入,包括基因表达数据的点对基因矩阵、相应组织切片的H&E图像以及将每个点的位置映射到图像上的X、Y坐标。
METI的目标是精确识别TME中各种细胞类型及其各自状态。METI的每个模块都针对特定细胞类型量身定制,利用领域特定知识进行专注分析(图. 1)。
Fig. 1 | Workflow of METI 分析
输入数据
- 10x Visium Spatial Transcriptomics (ST) 数据:包含基因表达数据的斑点-基因矩阵。
- Hematoxylin and Eosin (H&E) 图像:用于细胞形态学分析。
- XY坐标:将每个斑点的位置映射到图像上。
输出功能
- 细胞类型识别:识别并区分不同的细胞类型。
- 细胞核分割:对细胞核进行分割,以便进一步分析。
- 生成3D细胞密度图:可视化细胞在组织中的空间分布和密度。
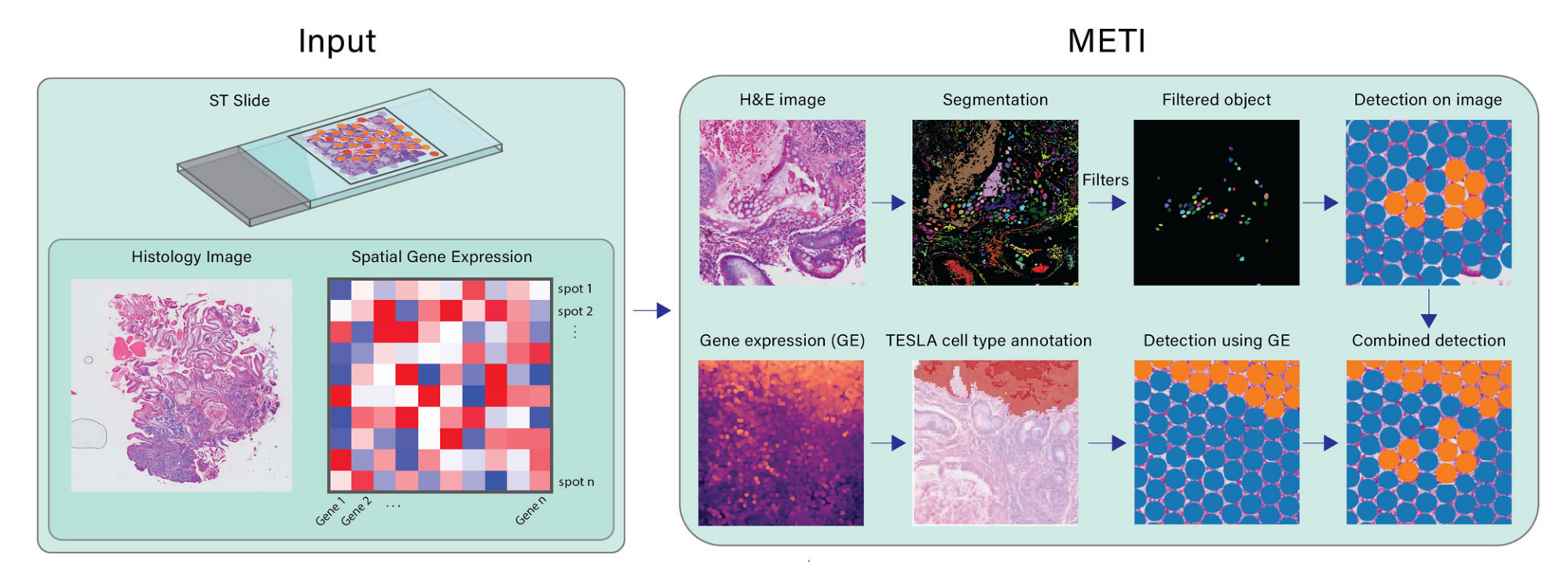
工作流程模块
-
模块1:正常和癌前细胞的映射
- 通过整合基因表达数据和H&E图像,识别和映射正常和癌前细胞,如胃中的杯状细胞。
- 具体步骤包括:
- 使用机器学习模型(如TESLA)结合元基因签名识别杯状细胞富集区域。
- 通过K-means分割方法在H&E图像中检测不同形态成分,进一步识别杯状细胞。
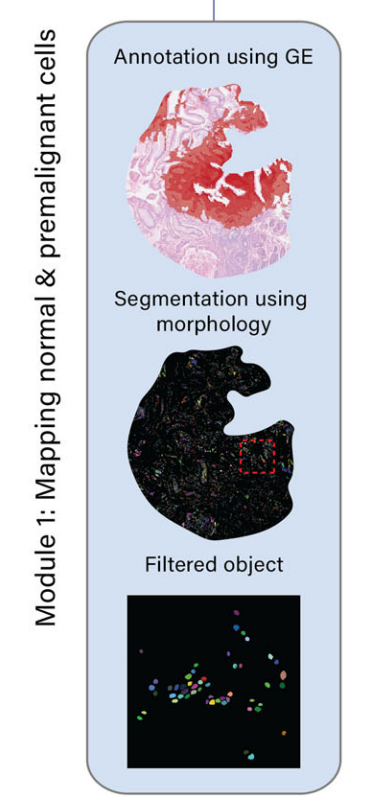
-
模块2:癌细胞领域的识别和异质性表征
- 使用癌细胞标记基因(如细胞角蛋白、EPCAM、三叶因子)识别肿瘤细胞富集区域。
- 通过额外的标记基因(如MKI67、SOX9、CLDN18、MSLN)表征癌细胞状态和异质性。
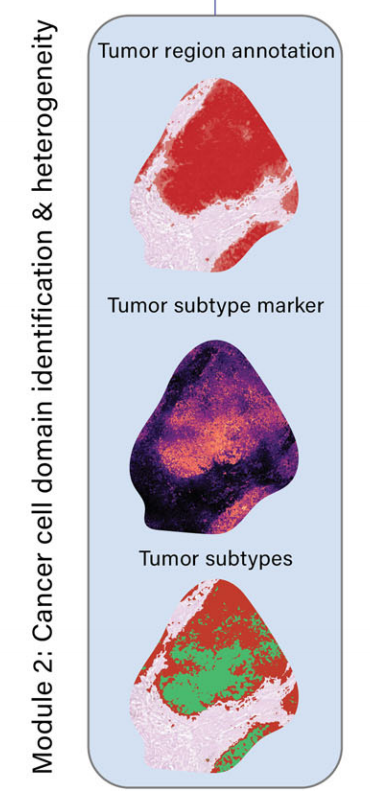
-
模块3:T细胞映射和表型分析
- 使用特定标记基因(如CD3D、CD3E、CD4、CD8A、CD8B)识别和映射T细胞富集区域。
- 进一步区分CD4+ T细胞、CD8+ T细胞及其不同状态(如调节性T细胞和耗竭T细胞)。
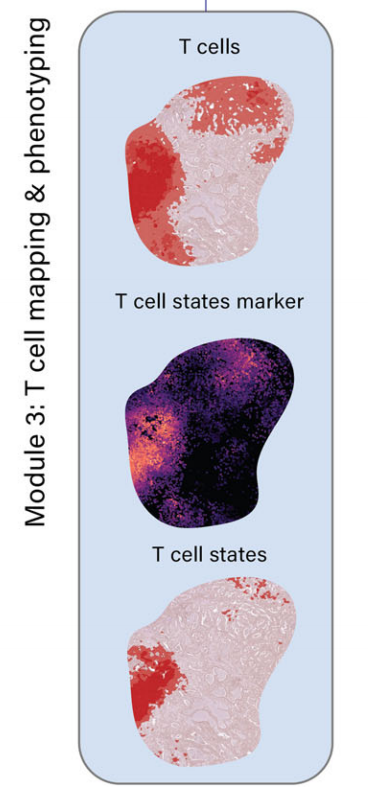
-
模块4:其他免疫细胞的深入分析
- 识别和分析除T细胞外的其他免疫细胞类型,如中性粒细胞、B细胞、浆细胞和巨噬细胞。
- 使用已验证的基因签名识别特定免疫细胞类型/状态。

-
模块5:癌症相关成纤维细胞(CAFs)的分析
- 识别和映射CAFs及其亚型(如myCAFs、iCAFs、apCAFs)。
- 通过特定的成纤维细胞标记基因识别和表征不同亚型的CAF。
在第一个模块中,METI识别正常和前恶性细胞,如胃部的杯状细胞17–19。模块2识别肿瘤细胞富集区域并表征其细胞状态异质性。模块3专注于T细胞的空间映射,包括CD4+和CD8+ T细胞,以及各种T细胞状态,如调节性T细胞(Treg)和耗竭性T细胞(Tex)。除了T细胞,模块4还识别其他免疫细胞,包括中性粒细胞、B细胞、浆细胞和巨噬细胞。在最后一个模块中,METI专注于对CAFs的综合分析,CAFs是一类在癌症进展和治疗抵抗中发挥关键作用的活化间质细胞20–22。该模块映射CAFs及其亚型,包括myCAFs、iCAFs和apCAFs23–27。
METI生成的输出结果全面,包括特定细胞类型的分割结果、基因表达数据以及提供组织样本整体视角的分割-基因表达整合结果,以及用于细胞密度空间可视化的3D密度图。
作者团队已经证明,与现有方法相比,METI在细胞类型识别方面更为准确。此外,即使在某一模态数据质量较低的情况下,METI也能保持稳健,因为高质量的数据来自另一模态可以补偿,确保作者团队分析的可靠性和有效性。
2-1:正常和前恶性细胞的映射
METI的第一个模块专注于解析上皮细胞室内的正常和前恶性细胞。
在这里,作者团队以杯状细胞为例,因为它们在H&E染色的图像中显示出独特的形态外观。杯状细胞呈酒杯状,顶部有淡色、几乎白色的囊泡,底部有椭圆形的细胞核。杯状细胞常见于呼吸道、消化道和生殖道,包括小肠、结肠和支气管。它们在维持这些组织的稳态中起着关键作用。在疾病的背景下,肠道中杯状细胞的异常存在是肠上皮化生这一前癌状态的关键特征17,19,28–30。
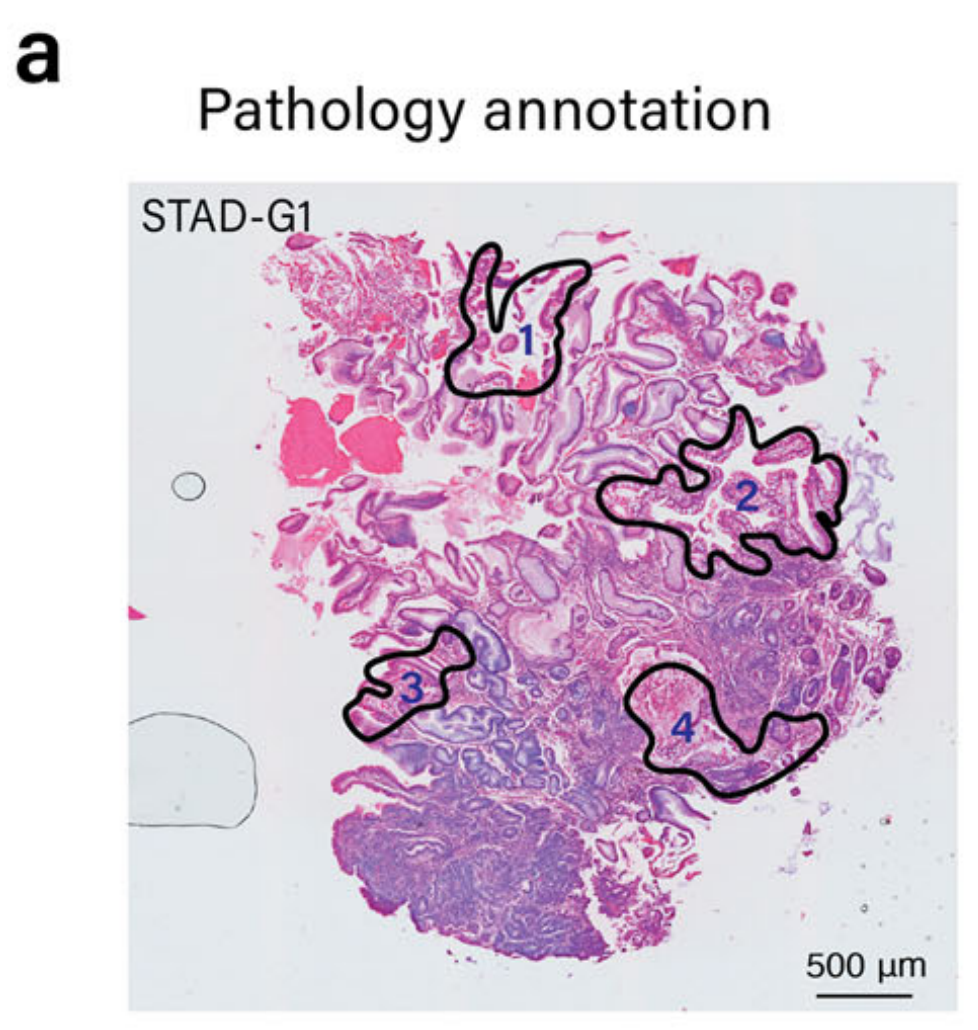
为了展示METI模块1的能力,作者团队将其应用于识别人类胃腺癌(STAD)样本中的杯状细胞,标记为G1,由作者团队的胃肠病病理学家注释(图. 2a)。
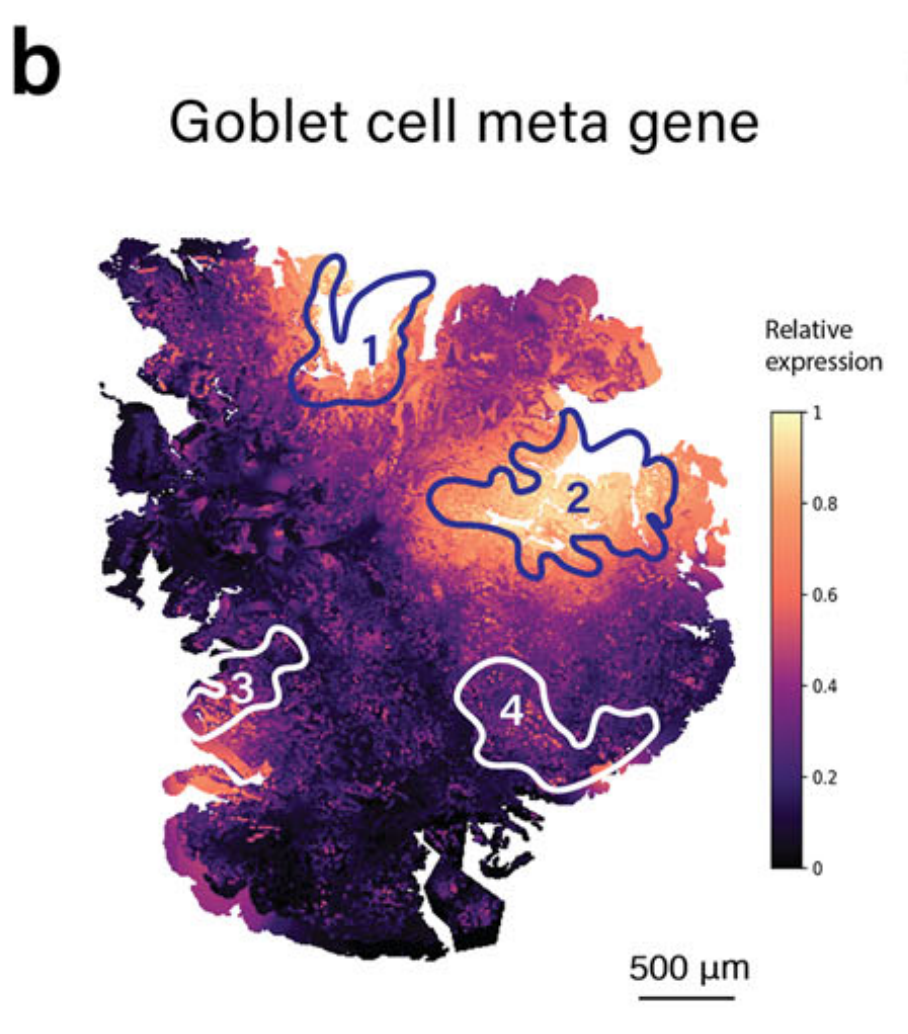
METI首先将先前出版物31–35中报道的典型杯状细胞标记物组合成一个元基因特征,包括MS4A10、MGAM、CYP4F2、XPNPEP2、SLC5A9、SLC13A2、SLC28A1、MEP1A、ABCG2和ACE2(补充表2)。
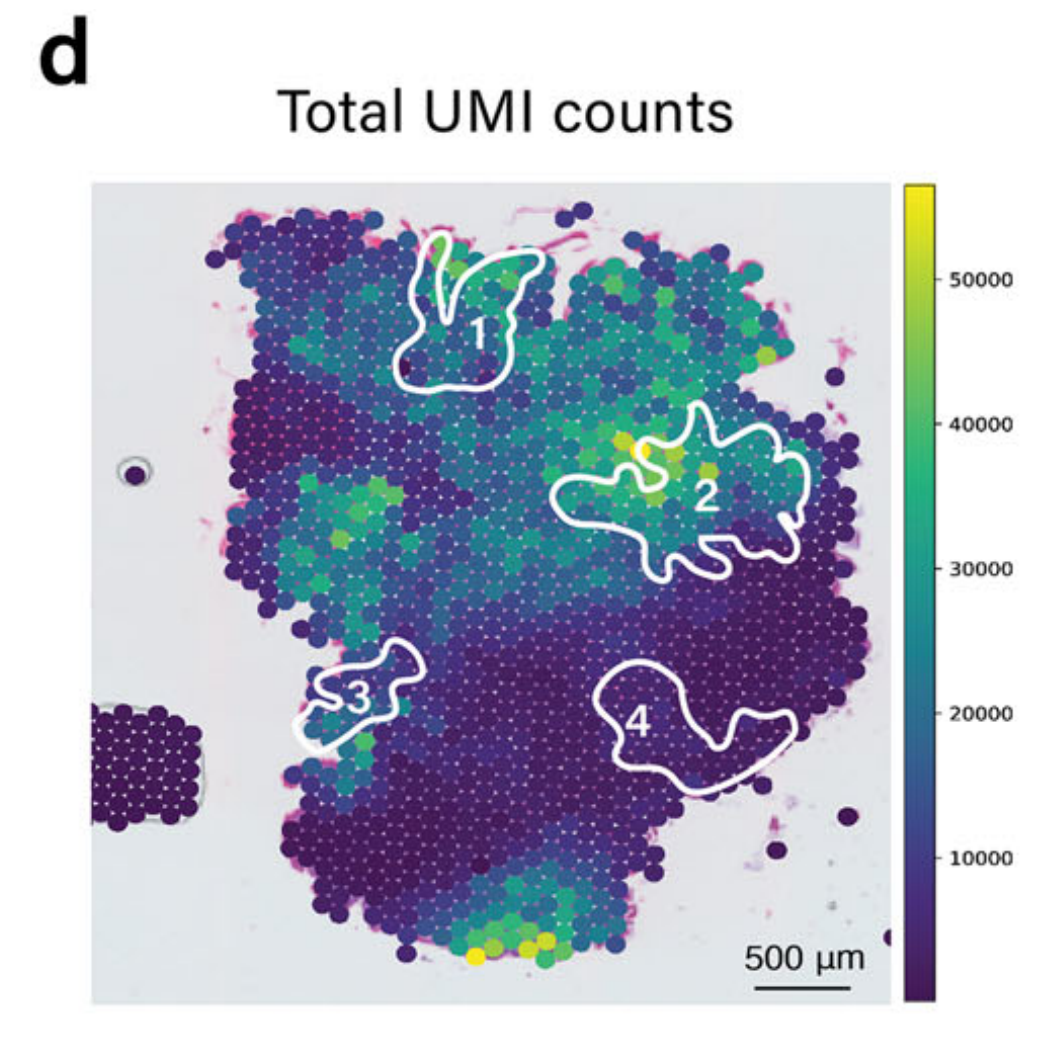
这个元基因在整张切片上可视化了杯状细胞分子特征的整体表达水平。如图2b所示,然后使用机器学习模型TESLA14,利用这个元基因来注释杯状细胞富集区域。
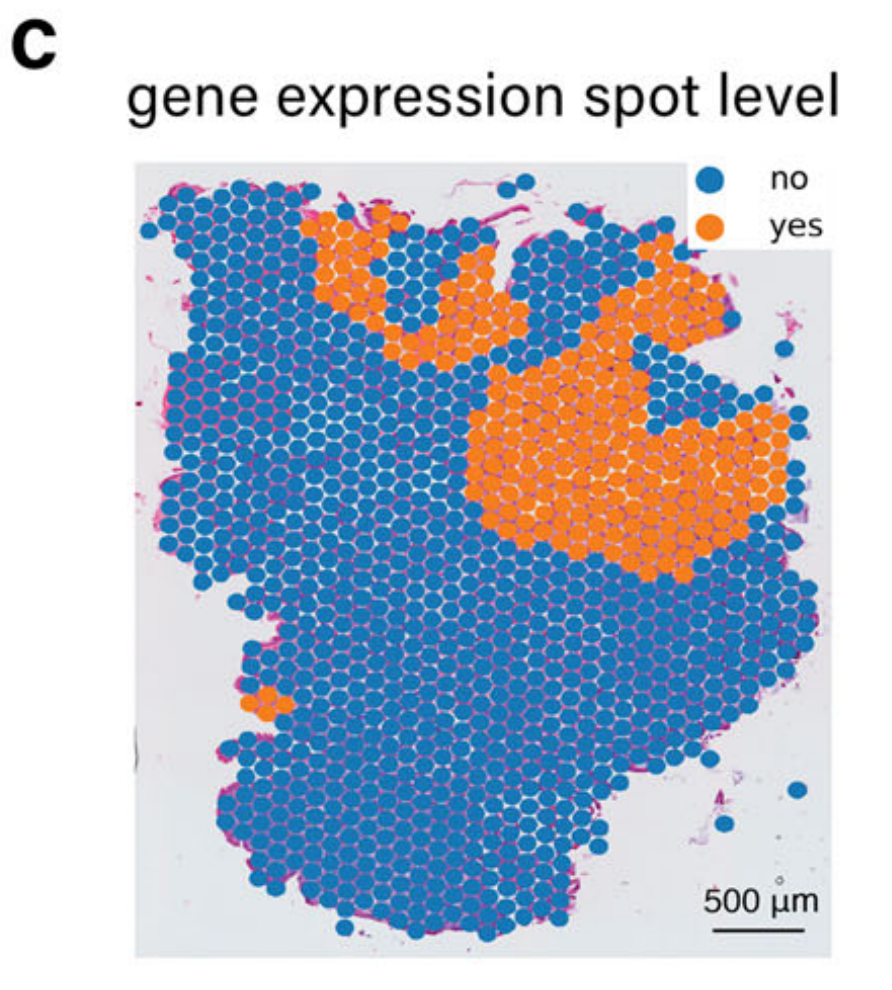
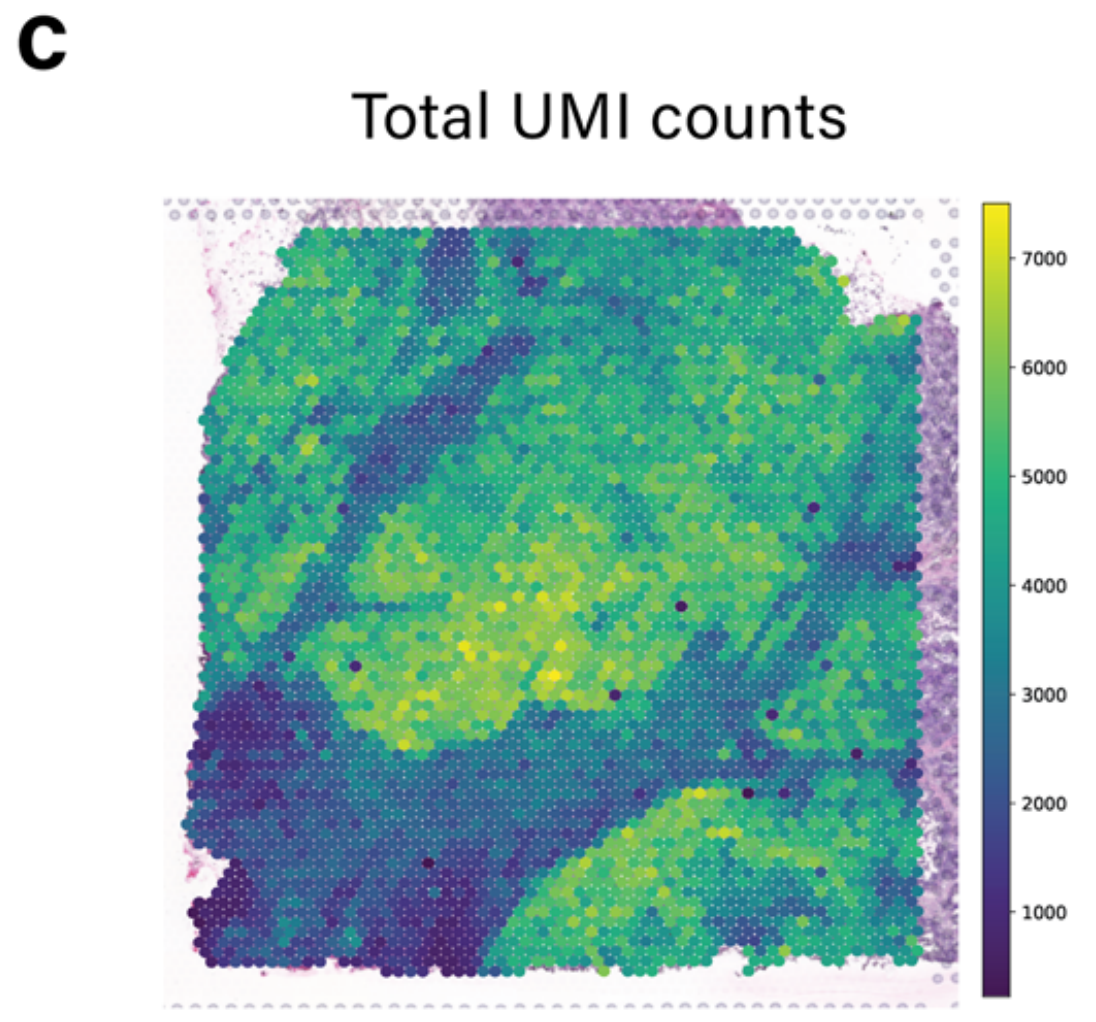
值得注意的是,TESLA中的细胞类型注释主要依赖于基因表达,由于区域变异和一些标记基因的高噪声水平,可能导致假阴性注释。例如,如图2c所示,TESLA未能识别区域4中的杯状细胞。
进一步检查显示,这种检测中的假阴性是由于区域4中捕获的独特分子标识符(UMI)计数整体较低,如图2d所示。
为了解决低质量基因表达数据造成的限制,METI同时也在H&E图像上执行杯状细胞的识别。METI采用基于K-means的分割方法来检测不同的形态学成分,如背景、细胞核、纤维、腺体和坏死。接下来,通过过滤这些成分的颜色、形状和大小(见方法),METI能够准确检测以圆形空心中心为特征的杯状细胞(图. 2e)。
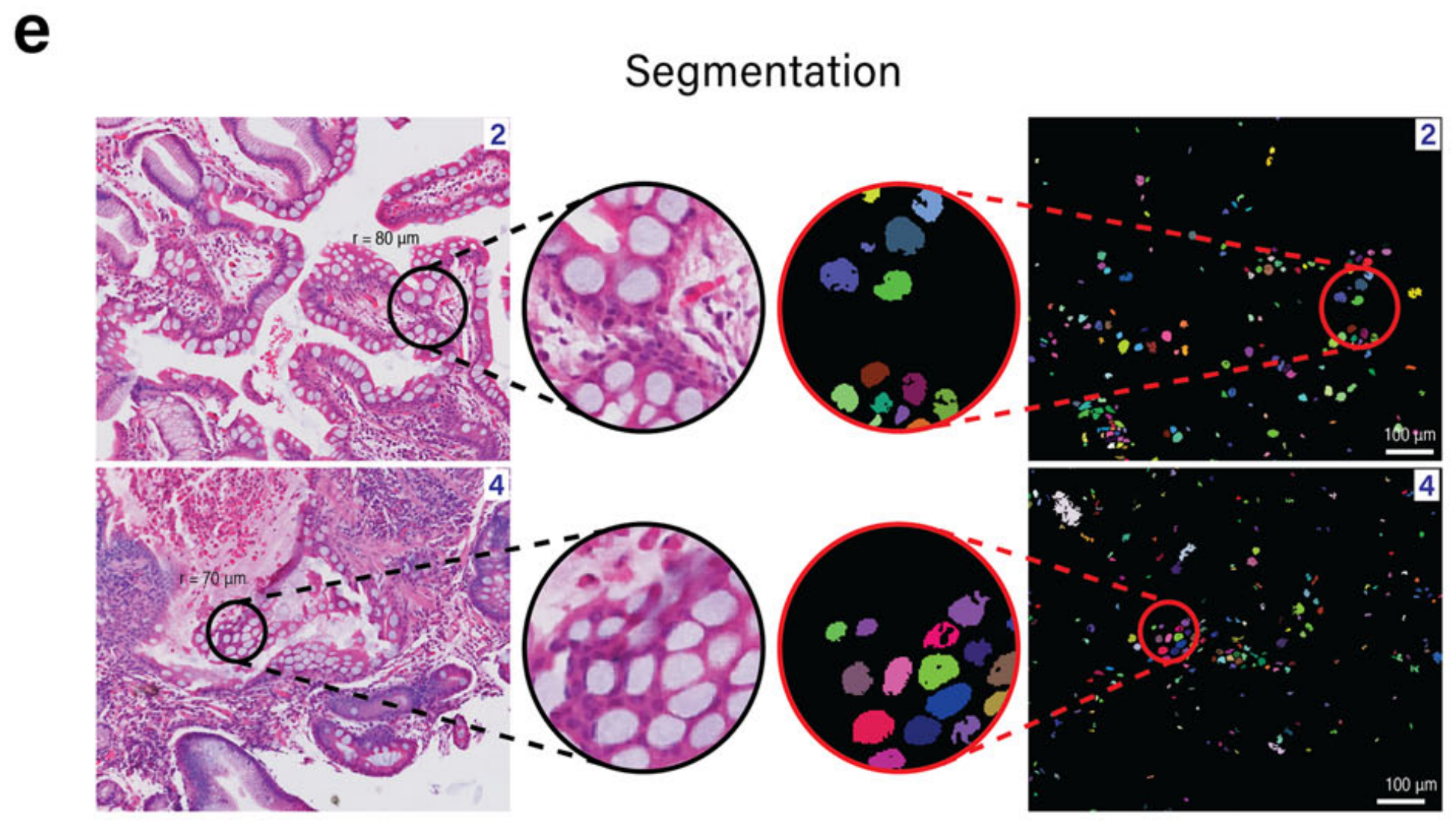
METI的这种形态学分析使得能够识别区域4中的杯状细胞(图. 2f),这是仅靠转录组数据所忽略的区域。
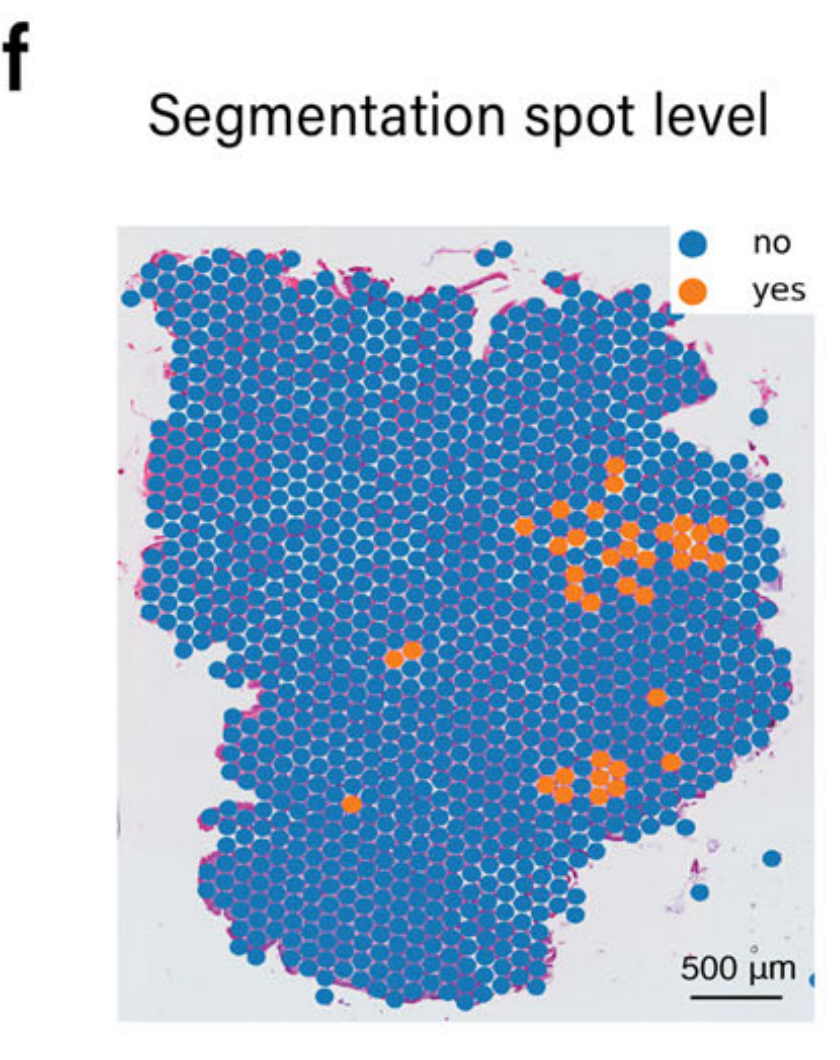
相反,由于组织碎片化,表现出不连续和脆弱的形状,区域1和3中的杯状细胞并未通过图像分析检测到。通过整合基因表达和图像分析结果,METI成功识别了所有四个富集杯状细胞的区域,如图2g所示。

这种整合方法克服了低UMI计数的限制,为分析样本中的杯状细胞提供了更准确的表征。利用基因表达数据和成像对杯状细胞检测的详细检查表明,只有通过这两种模态的整合才能实现准确的检测。
2-2:识别癌细胞区域及异质性
大多数实体肿瘤起源于上皮细胞,称为癌,包括胃癌、肺癌、膀胱癌、乳腺癌、前列腺癌和结肠癌,而其他一些实体肿瘤则起源于其他类型的组织,如肉瘤和黑色素瘤。
无论其起源细胞类型如何,理解恶性细胞的分子特征和细胞异质性对于揭示肿瘤生长、侵袭、转移和治疗反应的机制至关重要。因此,METI的第二个模块专注于恶性细胞的分析。
该模块首先使用作者筛选的癌细胞标记物,如细胞角蛋白(CK)、EPCAM和三叶因子来识别癌细胞。如图2h所示,METI有效地识别了STAD样本G2中的所有肿瘤区域,与经验丰富的病理学家的注释高度一致。

接下来,METI纳入额外的标记物来表征癌细胞的状态和异质性,包括细胞增殖标记物如MKI67来映射增殖性癌细胞,与干细胞相关的标记物如SOX9来识别STAD中的干细胞样癌细胞,以及治疗靶点如CLDN18和MSLN来进一步表征肿瘤亚型36–39。这些标记基因在样本G2的肿瘤区域中表现出独特的表达模式,如图2i所示。

它们可用于表征不同的癌细胞状态。例如,如图2j所示,METI不仅能识别SOX9+肿瘤区域,还能展示不同癌细胞状态在同一组织切片上的共定位或排他性。该模块提供了一个灵活且可定制的 approach,允许用户输入他们感兴趣的基因来进行肿瘤状态识别。

此外,用户可以利用与关键通路相关的基因,如KRAS、EGFR以及缺氧因子等,来全面探索不同癌症类型中的癌细胞状态和空间异质性。
量化生物组织中细胞的空间分布和密度对于多种应用至关重要,尤其是在病理学和肿瘤学领域。虽然基因表达提供了分子层面的视角,但相关的H&E图像可用于测量空间细胞分布和密度。遵循模块1中的并行过程,METI接下来进行了肿瘤细胞核的分割,并生成了3D肿瘤细胞密度图(图2k),直观地展示了癌细胞的空间分布和密度。

2-3:T细胞映射与表型分析
METI的模块3致力于表征TME中的T细胞及其各种状态。
最初,作者团队使用特定的T细胞标记物,包括CD3D和CD3E,来映射T细胞富集区域。在已识别的T细胞区域内,作者团队进一步区分T细胞的不同状态。
通过添加特定的细胞系标记物如CD4、CD8A和CD8B40,作者团队可以进一步区分CD4+ T细胞、CD8+ T细胞及其各种状态,包括CD4+ Tregs(如FOXP3、IL2RA)和CD8+ Tex细胞,通过纳入已知的免疫检查点基因(如PD-1、TIM-3、LAG-3、CTLA-4、TIGIT)和Tex相关转录因子(如TOX)40。
此外,该模块提供了在定义的癌细胞区域内叠加两种或多种不同T细胞状态的功能,允许作者团队可视化它们的空间关系。由于浸润T细胞的水平和空间分布是影响肿瘤免疫表型和免疫治疗反应的关键因素,METI的3D模块为整个图像创建了细胞密度图,用以直观展示T细胞在TME中的空间分布。
为了展示该模块的能力,作者团队应用METI分析了STAD样本G3和肺腺癌(LUAD)样本L1。
两个样本的病理学注释显示在补充图S2中。METI识别了在图3a中显示的T细胞基因表达水平升高的区域。
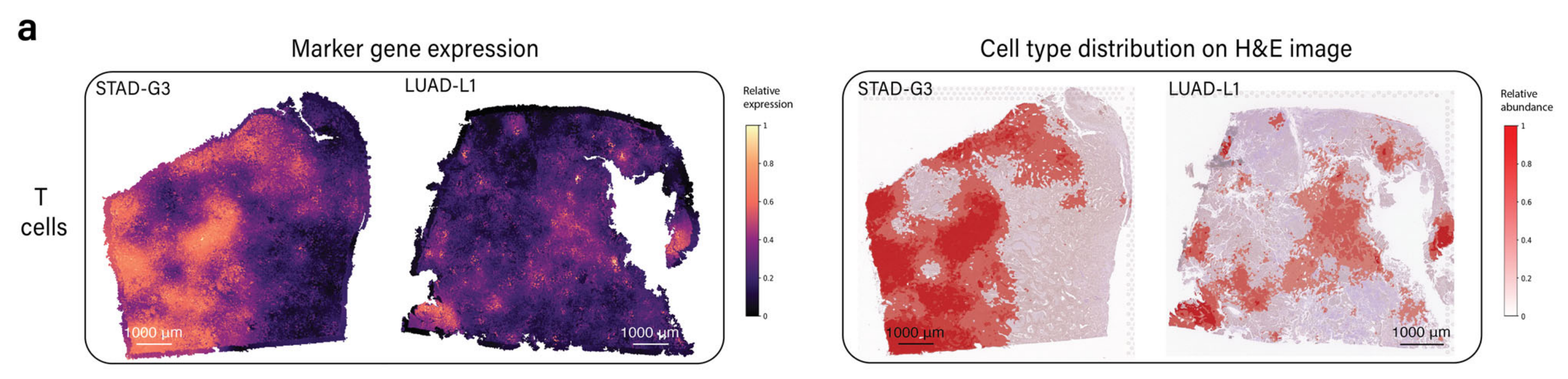
接下来,为了界定CD8+ T细胞富集区域,作者团队将分析限制在T细胞富集区域内。图3b显示了CD8+ T细胞富集区域,图3c、d显示了包括CD4+ Tregs和CD8+ Tex细胞的不同状态。

映射不同的T细胞状态有助于阐明在分析的STAD和LUAD样本中的空间景观和细胞间相互作用,促进生成有洞察力的假设。
由于CD4+ Tregs和CD8+ Tex细胞相对于癌细胞的位置影响肿瘤免疫表型41,42和免疫治疗反应,作者团队将癌细胞区域与CD4+ Tregs和CD8+ Tex细胞富集区域分别叠加,如图3e、f所示。

根据叠加结果,作者团队观察到不同样本中CD4+ Tregs和CD8+ Tex细胞的富集模式存在差异。具体来说,在样本G3中,CD4+ Tregs的数量略少于CD8+ Tex细胞。而在样本L1中,CD8+ Tex细胞的数量则少于CD4+ Tregs(图3e、f)。
这突显了不同肿瘤类型间T细胞状态的变异性。为了更好地展示整个图像的空间细胞分布,METI提供了基于H&E图像分割的细胞核密度的3D细胞密度图(图3g)。

对于STAD样本,左上角的一个区域显示较高的细胞密度,而LUAD样本则显示整个样本中相对均匀的细胞密度。
2-4:深入分析其他免疫细胞
METI的模块4能够检测T细胞以外的免疫细胞类型,包括中性粒细胞、巨噬细胞、B细胞和浆细胞,这些细胞是TME中的关键组成部分。
METI利用经过验证的基因签名来识别特定的免疫细胞类型/状态40,43–45。作者团队已将该模块应用于两个膀胱癌样本B1和B2的中性粒细胞检测。两个H&E图像显示在图4a和c中。
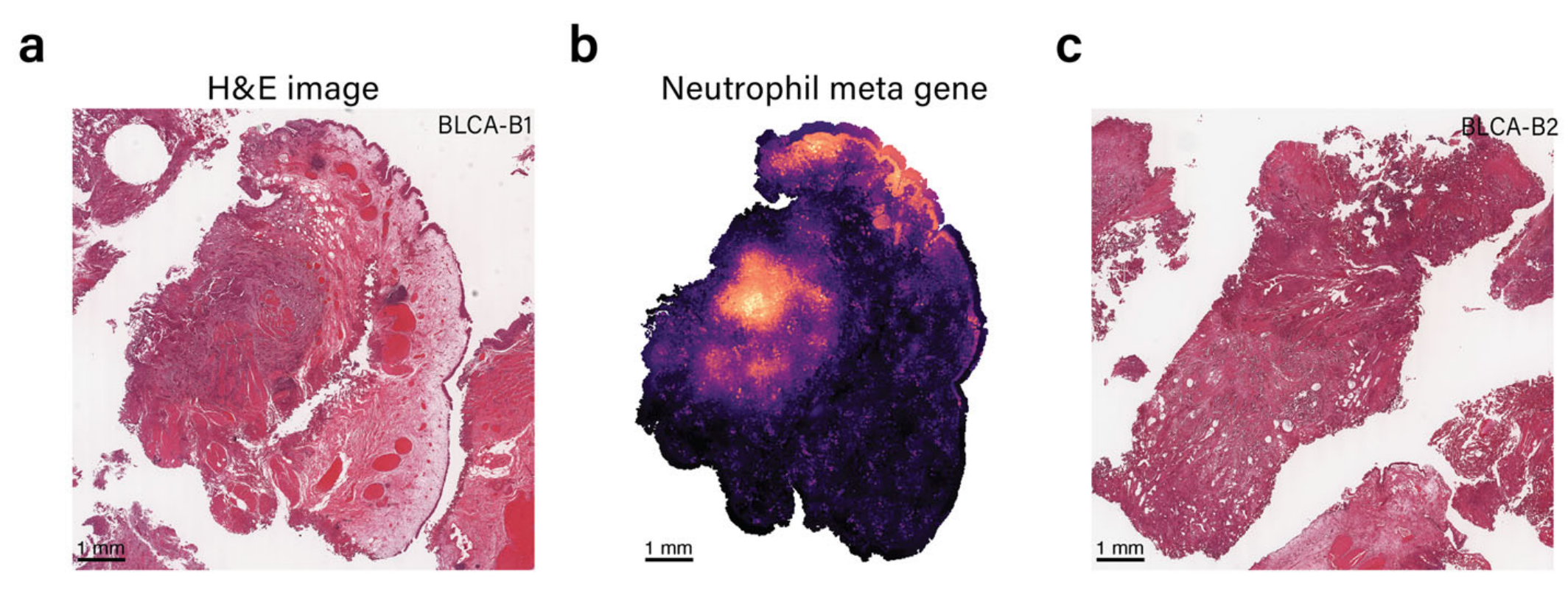
这些样本中的中性粒细胞富集区域由作者团队的经验丰富的病理学家验证为评估的地面真实(补充图S3)。如图4b和d所示,METI识别了两部分中表达中性粒细胞标记基因的区域。
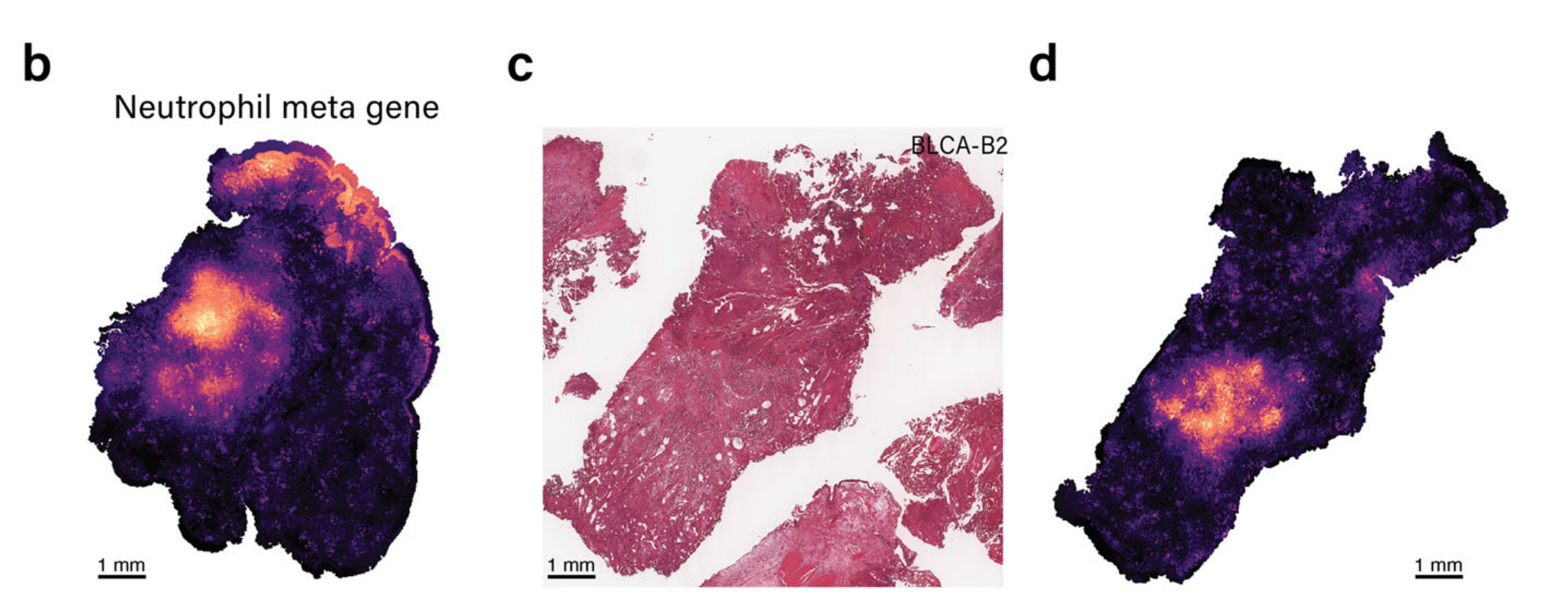
随后,METI直接在H&E图像上进行了相应的注释,隔离了表达中性粒细胞标记基因的区域,并提供了放大视图,如图4e所示。
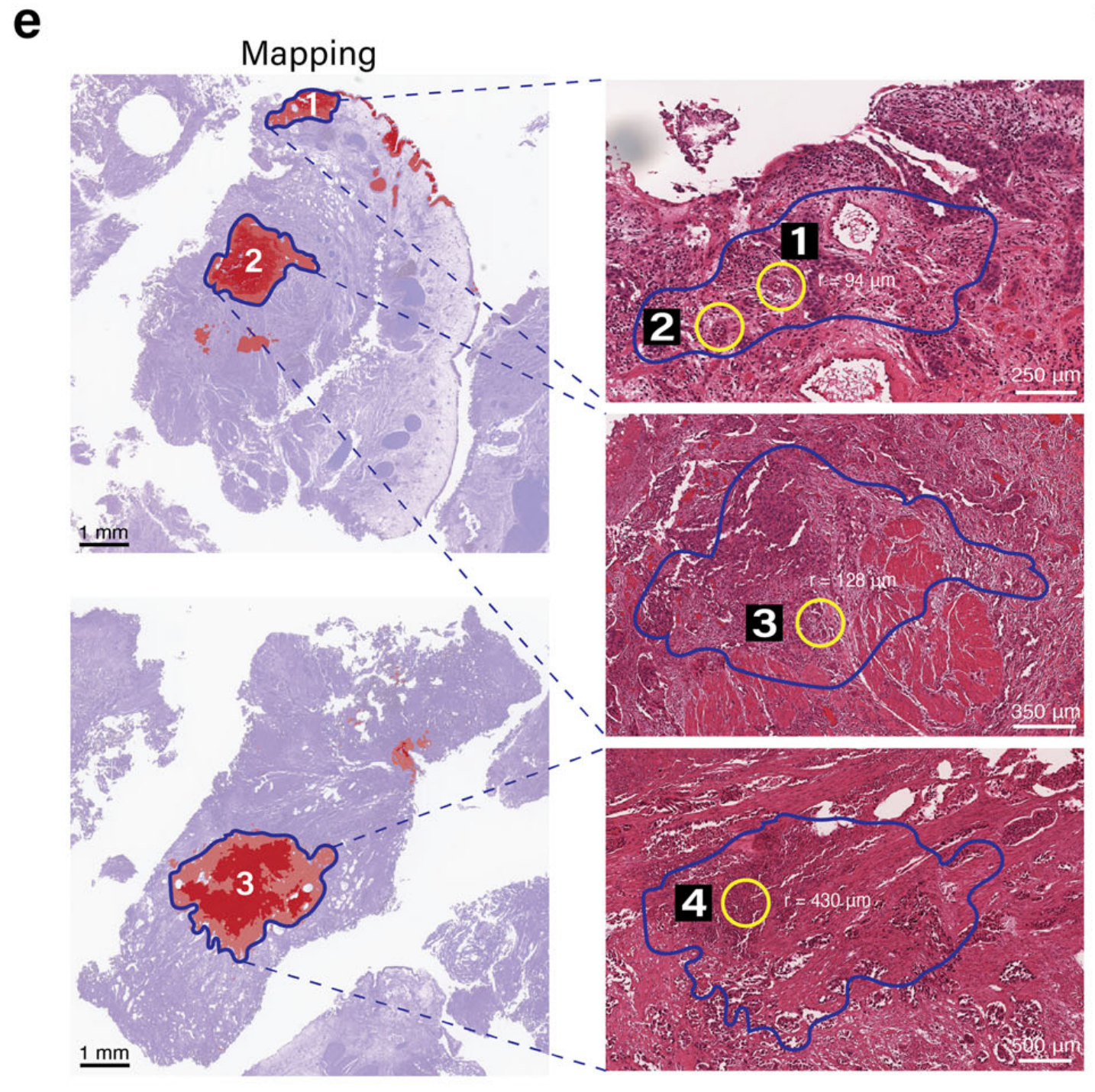
在放大这些注释区域后,表现出特征性的多叶核的中性粒细胞在图像分析中很容易区分。在图4f中,四个被病理学验证的中性粒细胞区域被圈出,随后进行了中性粒细胞检测。
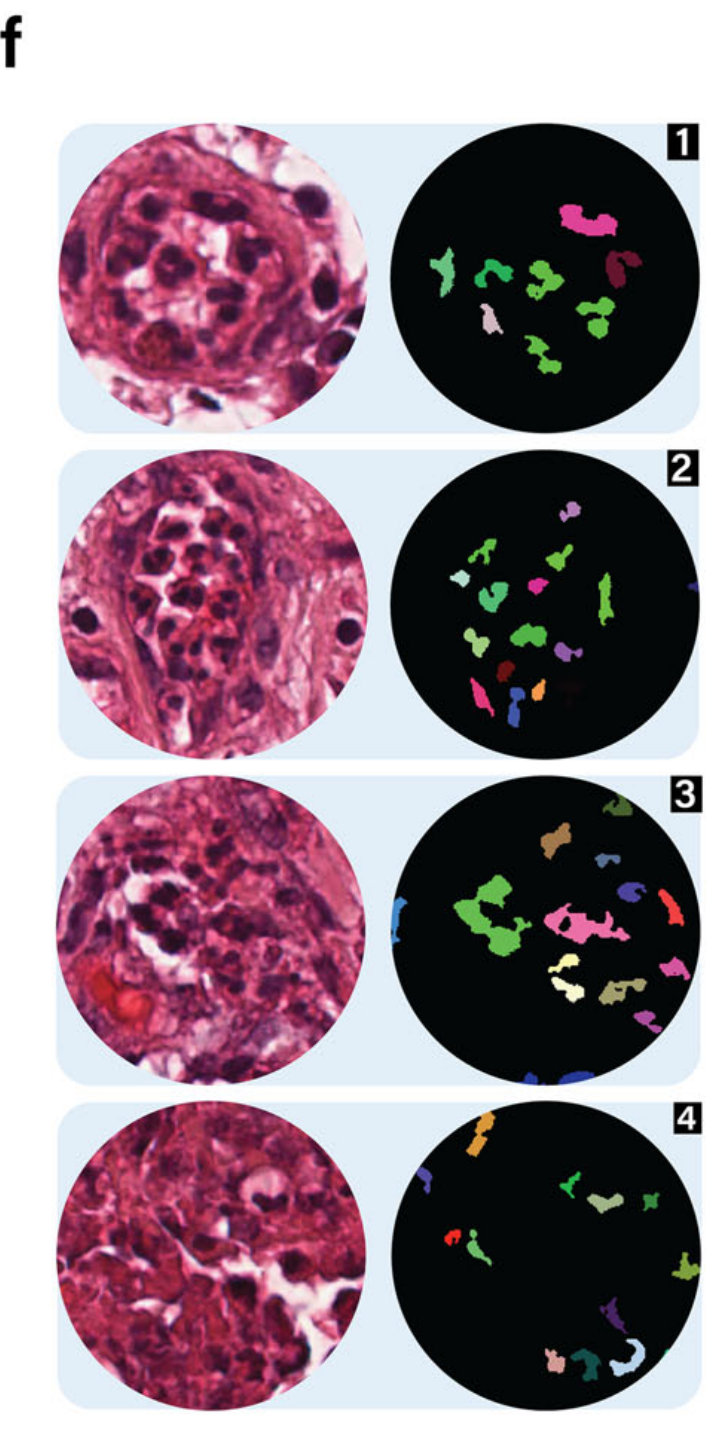
结果与使用基因表达的注释高度相关。作者团队还提供了如图4g所示的3D细胞密度图,以展示TME中中性粒细胞附近的空间细胞分布。

此外,作者团队还展示了该模块的能力,通过映射STAD样本中的B细胞和浆细胞(图4h和i)。
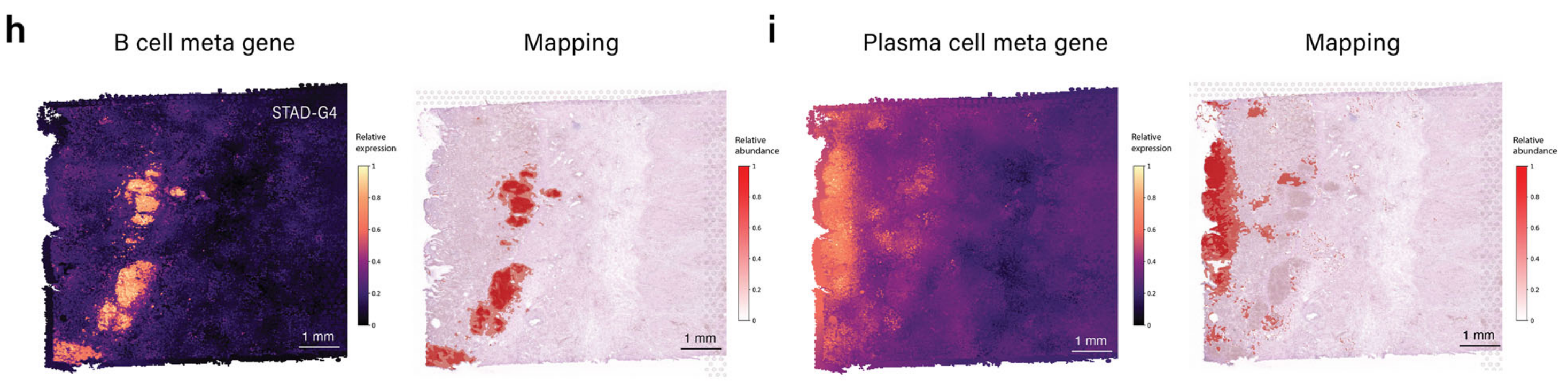
3D细胞密度图(图4j)与作者团队的病理学家标注的STAD样本G4中的淋巴样聚集物(补充图S2a)很好地对齐。

同样,根据病理学注释,可以在LUAD样本中正确映射巨噬细胞(图4k和补充图S2b)。
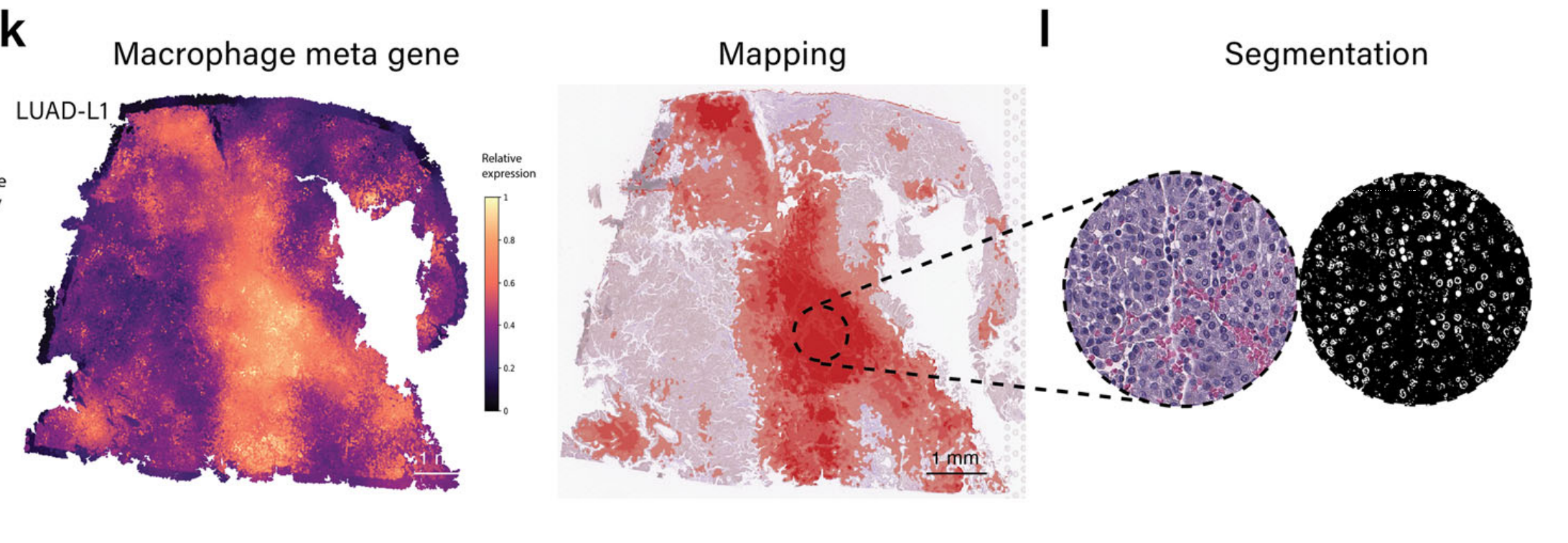
在显示高巨噬细胞标记基因表达的区域中,随机选择了区域进行分割,揭示了一群巨噬细胞(图4l)。除了上述免疫细胞类型外,该模块保持灵活性,允许用户使用他们编写的基因签名来调查其他感兴趣的特定免疫细胞群体。
2-5:分析与癌症相关的成纤维细胞(CAFs)
在模块5中,METI被设计用来分析TME中的基质细胞成分,包括CAFs及其各种CAFs亚型。
CAFs以其显著的表型和功能异质性而闻名46–48。它们被归类为活化的成纤维细胞,代表TME中具有肿瘤促进和肿瘤抑制活动的关键成分49–51。
CAFs在表型和功能上都是异质的。已经识别和描述了不同类型的CAFs,如肌成纤维细胞样CAFs(myCAFs)、炎性CAFs(iCAFs)和抗原呈递CAFs(apCAFs)23–27。
作者团队将模块5应用于被作者团队的病理学家注释为含有丰富肿瘤间质的胃样本G2(图5a)。
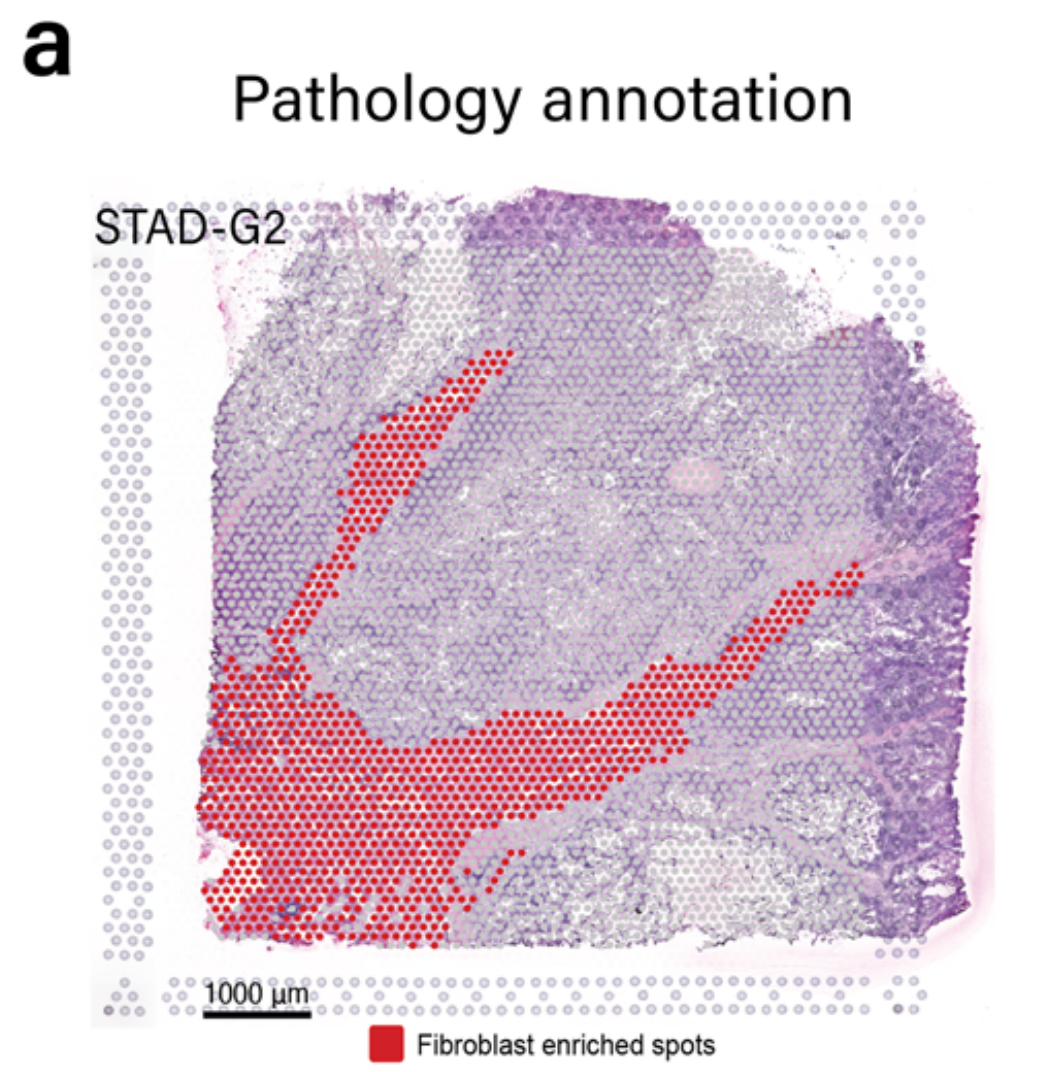
METI首先分割CAFs,并生成了如图5b所示的成纤维细胞细胞密度图。

接下来,作者团队发现METI注释的富集纤维细胞区域具有减少的UMI计数,这与癌细胞倾向于具有较高的UMI计数相比其他细胞类型的观点相一致(图5c)。
METI接着有效地在样本中映射CAFs,并使用CAF元基因(图5d)在H&E图像上注释CAFs,这与病理学注释高度一致。

在注释的CAF区域内,METI进一步深入研究CAF亚型的表征,包括myCAFs、iCAFs和apCAFs(图5e–g)。为了表征CAF亚型的空间共定位,作者团队在这三种CAF群体上叠加了总CAF阳性区域(图5h)。

这种方法使作者团队能够更好地理解TME中CAFs的空间异质性。同样,METI可以共映射CAFs、癌细胞和其他感兴趣的免疫细胞亚集,以提供有关它们之间细胞相互作用的额外见解。该模块保持适应性,允许用户根据他们的特定兴趣探索与CAFs相关的其他子区域。
2-6:定量比较与现有工具
作者团队首先比较了METI与两种空间聚类方法SpaGCN13和BayesSpace12的性能,特别是在人类STAD数据集中对杯状细胞注释的背景下。病理学家的注释作为基准,指示了H&E图像上杯状细胞的位置,如图S4a、b所示。METI在识别这些斑点方面表现出高准确性(ACC = 0.778),如图S4c所示。
为了与SpaGCN和BayesSpace的无监督性质进行公平比较,作者团队建立了一个标准:如果一个聚类中的超过10%的斑点是真正的杯状细胞,那么这个聚类就被认为是杯状聚类。这种方法允许作者团队将聚类分为杯状和非杯状组进行二元比较,并计算BayesSpace和SpaGCN的准确性。BayesSpace的聚类结果如图S4d所示。
BayesSpace在n = 15聚类时达到最高准确度0.704,这低于METI。进一步使用河流图分析,如图S4e所示,BayesSpace无法隔离一个完全由杯状细胞组成的单一聚类,无论选择的聚类数量如何。SpaGCN也遇到了类似的问题,如图S4f所示,其最高准确度为0.734,在n = 5聚类时。这些发现强调了METI在细胞类型注释方面相对于现有聚类方法的优越性。
尽管METI和空间聚类方法都是参考无关的,但METI能够通过将其框架中的领域知识融入其中,提供准确的细胞类型注释。相比之下,空间聚类方法主要捕获整体组织结构,而不是细胞类型富集。
此外,作者团队利用胃单细胞RNA测序数据作为参考,评估了METI、RCTD和CytoSpace在识别两个不同的人类胃癌空间转录组(ST)数据集中的杯状细胞、CD4+ T细胞和CD8+ T细胞的表现,如图S5所示。
对于第一个数据集,RCTD在图S5a中显示出有限的准确性0.525,在准确确定杯状细胞位置方面明显遇到困难。相比之下,CytoSpace在图S5b中显示出显著更高的准确性0.751,与METI在图S5c中的发现一致。
由于第二个数据集中缺乏病理学注释,因此无法量化准确性,作者团队专注于比较RCTD和CytoSpace在识别CD4+ T细胞和CD8+ T细胞方面的能力。RCTD在准确识别CD4+ T细胞和CD8+ T细胞方面能力有限(图S5d、g),而CytoSpace在图S5e、h中优于RCTD。
因此,为了进一步验证这些发现,作者团队分析了标记基因的表达,以视觉评估结果,如图S5f、i所示,这进一步支持了METI和CytoSpace识别的细胞类型位置的存在。
2-7:METI在低质量图像中的稳健性
作者团队使用另一个人类STAD数据集来展示METI在低质量图像中的稳健性。
如图S6a所示,由于摄像机焦点的丢失导致的模糊性掩盖了细胞边界,使得作者团队的病理学家难以提供淋巴细胞的详细注释,因为不同区域之间的边界粗糙,如图S6b所示。在这个数据集中,作者团队应用METI进行B细胞注释。B细胞与其他淋巴细胞在H&E图像中通过它们的小、深紫色细胞核可区分。
因此,METI的初步步骤是识别H&E图像上的淋巴细胞。如图S6c所示,图像的模糊性导致大量假阳性淋巴细胞检测,这些细胞稀疏分布,特别是在组织边缘附近。
此外,H&E染色无法充分区分B细胞与其他淋巴细胞,因为它们的形态特征相似。METI的后续步骤是使用特定的基因标记,包括MS4A1和CD19,来识别B细胞。图S6d展示了仅通过基因表达识别的B细胞分布。由于B细胞是淋巴细胞的一个特定亚型,METI进一步通过将B细胞数据与从图像分析中识别的淋巴细胞区域叠加,以知识感知的方式精炼检测,如图S6e所示。
结果随后转换为斑点级数据,如图S6f所示。这个案例展示了METI能够以知识感知的方式合并基因表达和图像分析,即使在图像质量较低的情况下,也能确保细胞类型注释的稳健性。
三、讨论
在本研究中,作者团队介绍了METI,这是一个稳健的机器学习框架,旨在满足对TME中多种细胞类型及其表型状态进行全面表征的需求。
通过有效地整合转录组数据与组织病理学图像信息,METI减少了在TME中关键细胞类型非特异性映射的风险。与某些现有方法不同,其性能不依赖于其参考数据中细胞类型注释的质量与粒度,METI在这些因素上不受限制,为各种数据集提供了相当的灵活性和适用性。这些特性使METI与现有的细胞去卷积工具区分开来,后者主要依赖于基因表达数据,而忽略了组织病理学。
虽然METI利用了如TESLA和K-means分割等已建立的工具作为其分析框架,但这些方法最初作为通用工具开发,并未专门针对癌症ST数据分析进行优化。METI的创新之处在于其能够以知识驱动的方式补偿一个模态中低质量数据的问题,通过稳健地整合组织形态学与转录组学特征。作者团队已系统地对METI与多种流行的空间聚类和细胞类型去卷积方法进行了基准测试,结果表明METI在分析TME的关键组成部分时具有较高的准确性、高分辨率以及改进的可视化能力。
METI包含五个模块,它们表征了各种细胞组分,包括肿瘤细胞、免疫细胞和间质细胞的独特类型和状态。通过联合分析转录组学和组织病理学信息,METI提供了一个比单独分析基因表达和病理学图像更全面的方法来理解癌细胞和TME。
值得注意的是,作者团队的结果表明,METI能够通过利用另一个模态的高质量信息来减轻一个模态中低质量数据的影响。如在杯状细胞检测案例中,METI的整合结果弥补了低质量的基因表达数据。此外,这种整合方法使METI对H&E图像的低分辨率或伪迹问题具有稳健性。
此外,METI独特地能够分层各种细胞状态,包括CD4+ Tregs、CD8+ Tex细胞、iCAFs、myCAFs、apCAFs等。值得注意的是,这种分层不仅限于上述亚型;用户可以根据研究需求定义和探索其他细胞亚型。METI被设计为用户友好和易于访问。
为此,作者团队在METI框架内集成了预定义的细胞类型标记和模型参数。根据最近的泛癌研究40,52,免疫和间质细胞的细胞系标记和细胞表型状态标记的表达通常在不同肿瘤组织和数据集中是一致的,这为METI提供了有价值的参考。
因此,METI可以有效使用,无需用户指定输入,使其成为不同水平的生物信息学专家都可使用的工具。此外,考虑到研究需求的多样性和不同组织类型和疾病的特殊性,METI为更高级的用户提供了灵活性。研究人员可以通过输入他们感兴趣的标记,根据特定组织或疾病进行定制,以进行高度专业化的分析。通过结合预定义的标记和用户引入标记的能力,METI在易用性和可定制性之间架起了桥梁,满足了广泛的研究需求。
METI具有潜在的转化价值和实际用途。通过无缝集成到现有的临床诊断和治疗规划工具中,METI有可能补充和增强当前的诊断工作流程,促进临床环境中的更明智决策。尽管METI为深入的癌症细胞和TME细胞表征提供了一个有用的框架,但重要的是要认识到其局限性。
一个显著的挑战源于对转录组数据的依赖,这可能受到技术波动、样本质量限制、丢失发生以及依赖于用户提供的基因签名的影响,可能导致产生假阴性结果。此外,METI的分割功能性能可能会因被分析图像的质量与分辨率而有所不同。