单细胞空间转录组分析流程学习python版(三)
既往推文:
单细胞空间转录组分析流程学习(一):https://mp.weixin.qq.com/s/E4WuPokBOxKRbBF6CEB5aA
单细胞空间转录组分析流程学习(二):https://mp.weixin.qq.com/s/8AFeq50Dc91cI_6jdQZfFg
分析步骤
1.配置一个环境/安装必要软件
conda creadt -n scRNA_spatial python=3.9
conda install -c conda-forge scanpy pandas matplotlib seaborn
2.导入
import scanpy as sc
import anndata as an
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import seaborn as sns
sc.logging.print_versions()
sc.set_figure_params(facecolor="white", figsize=(8, 8))
sc.settings.verbosity = 3
3.读入文件
adata = sc.read_visium(path = "./RawData/GSE6716963",
count_file="GSM6716963_19G081_filtered_feature_bc_matrix.h5")
4.check
adata
#AnnData object with n_obs × n_vars = 2887 × 17943
# obs: 'in_tissue', 'array_row', 'array_col'
# var: 'gene_ids', 'feature_types', 'genome'
# uns: 'spatial'
# obsm: 'spatial'
再次学习一下这里的参数含义:
n_obs × n_vars:2887 个观测点(通常是细胞或空间点)和 17943 个基因。
obs:存储每个观测点的元数据,如是否位于组织中以及在空间阵列上的坐标。
var:存储每个基因的元数据,如基因 ID、基因类型和基因组信息。
uns:存放未结构化数据,如与空间信息相关的元数据。
obsm:存储每个观测点的多维数据(如空间坐标或降维结果)。
adata.obsm['spatial']
# array([[3426, 1363],
# [3980, 4005],
# [1430, 1658],
# ...,
# [4028, 3652],
# [3196, 3765],
# [1068, 3040]])
这表示每个观测点在组织切片中的位置。每一行是一个观测点的坐标,其中第一个数是 x 坐标,第二个数是 y 坐标。
adata.obsm['spatial'].shape
# (2887, 2)
2887:这个矩阵有 2887 行,表示有 2887 个观测点(细胞或空间点)。
2:这个矩阵有 2 列,表示每个观测点有 2 个空间坐标(通常是二维的 x 和 y 坐标)。
adata.uns['spatial'].keys()
# dict_keys(['19G081'])
adata.uns['spatial'].keys() 返回了字典中键的名称 '19G081',这是一个样本或切片的标识符
adata.uns['spatial']['19G081'].keys()
# dict_keys(['images', 'scalefactors', 'metadata'])
表示19G081字典下面储存着三项内容:images/scalefactors/metadata
adata.uns['spatial']['19G081']['images'].keys()
# dict_keys(['hires', 'lowres'])
iamges下面有高低分辨率两种图片
adata.uns['spatial']['19G081']['images']['hires'].shape
# (1878, 2000, 3)
adata.uns['spatial']['19G081']['images']['lowres'].shape
# (564, 600, 3)
adata.uns['spatial']['19G081']['scalefactors']
#{'tissue_hires_scalef': 0.36231884,
# 'tissue_lowres_scalef': 0.10869565,
# 'fiducial_diameter_fullres': 67.115571765934,
# 'spot_diameter_fullres': 41.547734902721054}
tissue_hires_scalef 和 tissue_lowres_scalef:用于将图像的像素坐标转换为实际组织坐标,分别对应高分辨率和低分辨率图像。
fiducial_diameter_fullres:在全分辨率图像中,标识性点(fiducial marker)的直径,通常用于图像校准。
spot_diameter_fullres:在全分辨率图像中,空间点(spot)的直径,表示测量基因表达的区域的实际大小。
adata.uns['spatial']['19G081']['metadata']
#{'chemistry_description': "Spatial 3' v1",
# 'software_version': 'spaceranger-1.3.0'}
adata.obs.head()
这里跟seurat中的很相似,tissue代表不同的组织切片/row和col代表位置坐标
5.数据质量控制
adata.var_names_make_unique()
adata.var["mt"] = adata.var_names.str.startswith("MT-")
sc.pp.calculate_qc_metrics(adata, qc_vars=["mt"], inplace=True)
adata.var_names_make_unique() 确保基因名称唯一。
adata.var["mt"] = adata.var_names.str.startswith("MT-") 标记线粒体基因。
sc.pp.calculate_qc_metrics 计算质量控制指标,包括线粒体基因表达的比例,用于后续数据过滤和质量评估。
adata.obs.head()
6.增加slice列
adata.obs['slice'] = ['slice' + str(i) for i in adata.obs['in_tissue']]
adata.obs.head()
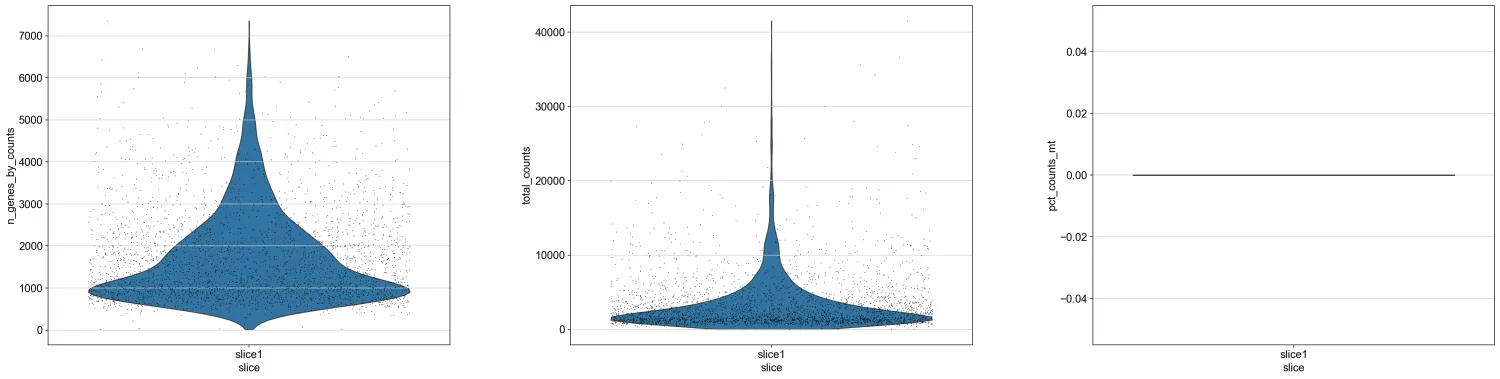
7.绘制一下count的小提琴图
sc.pl.violin(adata, ['n_genes_by_counts', 'total_counts', 'pct_counts_mt'], groupby="slice", jitter=0.4, multi_panel=True)
8.过滤数据
也有老师说可以不过滤hhhh
sc.pp.filter_cells(adata, min_counts=5000)
sc.pp.filter_cells(adata, max_counts=35000)
adata = adata[adata.obs["pct_counts_mt"] < 20]
sc.pp.filter_genes(adata, min_cells=10)
9.归一化/高变基因
sc.pp.normalize_total(adata, inplace=True)
sc.pp.log1p(adata)
sc.pp.highly_variable_genes(adata, flavor="seurat", n_top_genes=2000)
10.降维聚类
sc.pp.pca(adata)
sc.pp.neighbors(adata)
sc.tl.umap(adata)
sc.tl.leiden(adata, key_added="clusters")
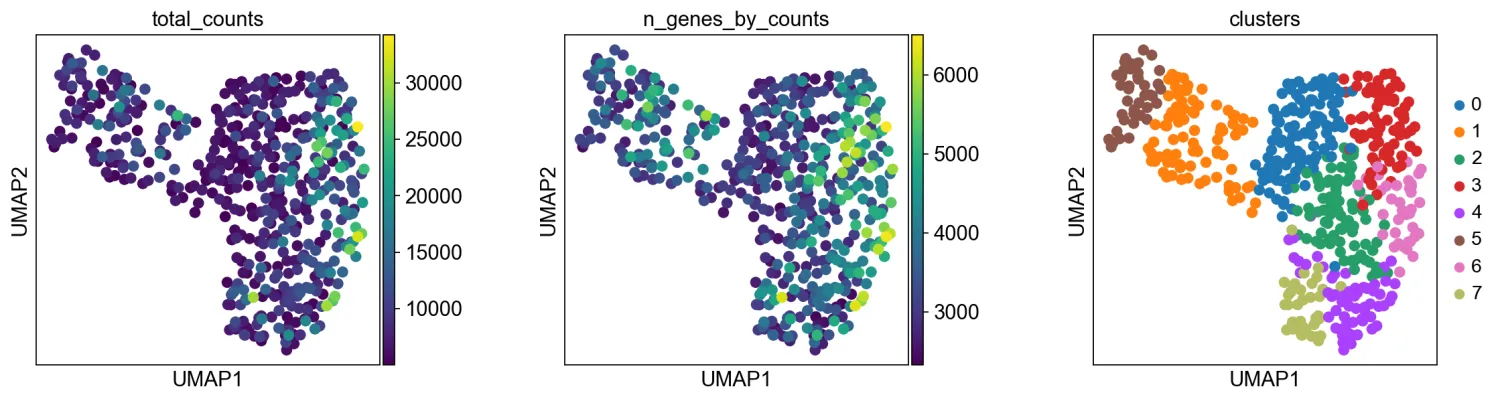
11.UMPA绘图
plt.rcParams["figure.figsize"] = (4, 4)
sc.pl.umap(adata, color=["total_counts", "n_genes_by_counts", "clusters"], wspace=0.4)
12.空转绘图
plt.rcParams["figure.figsize"] = (8, 8)
sc.pl.spatial(adata, img_key="hires", color=["total_counts", "n_genes_by_counts", "clusters"], alpha_img=1)
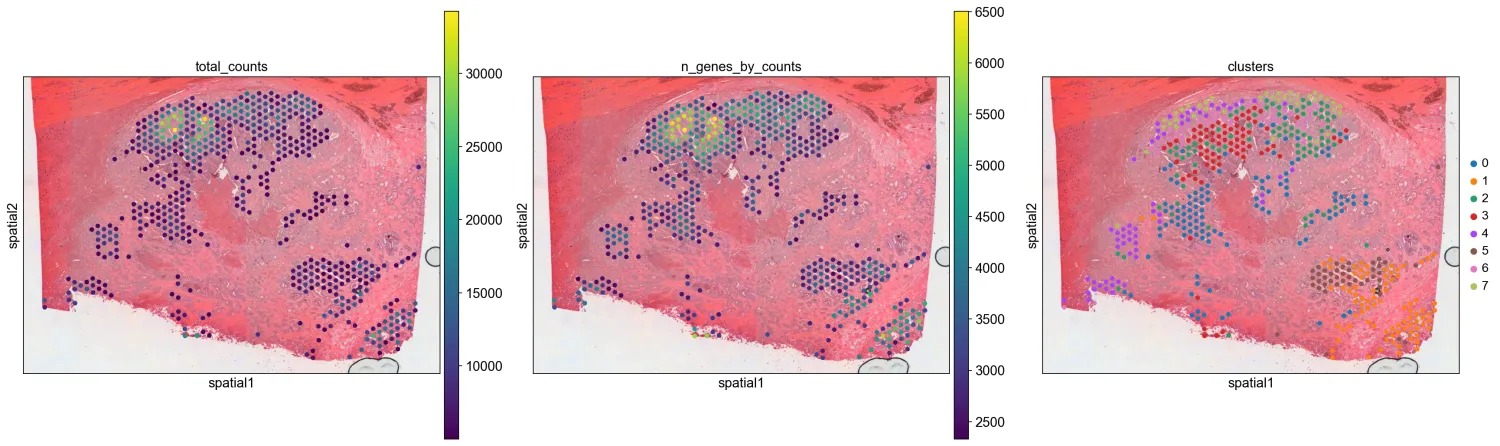
sc.pl.spatial(adata, img_key="hires", color="clusters", groups=["1","2", "3"], size = 1.3)
特定cluster绘制

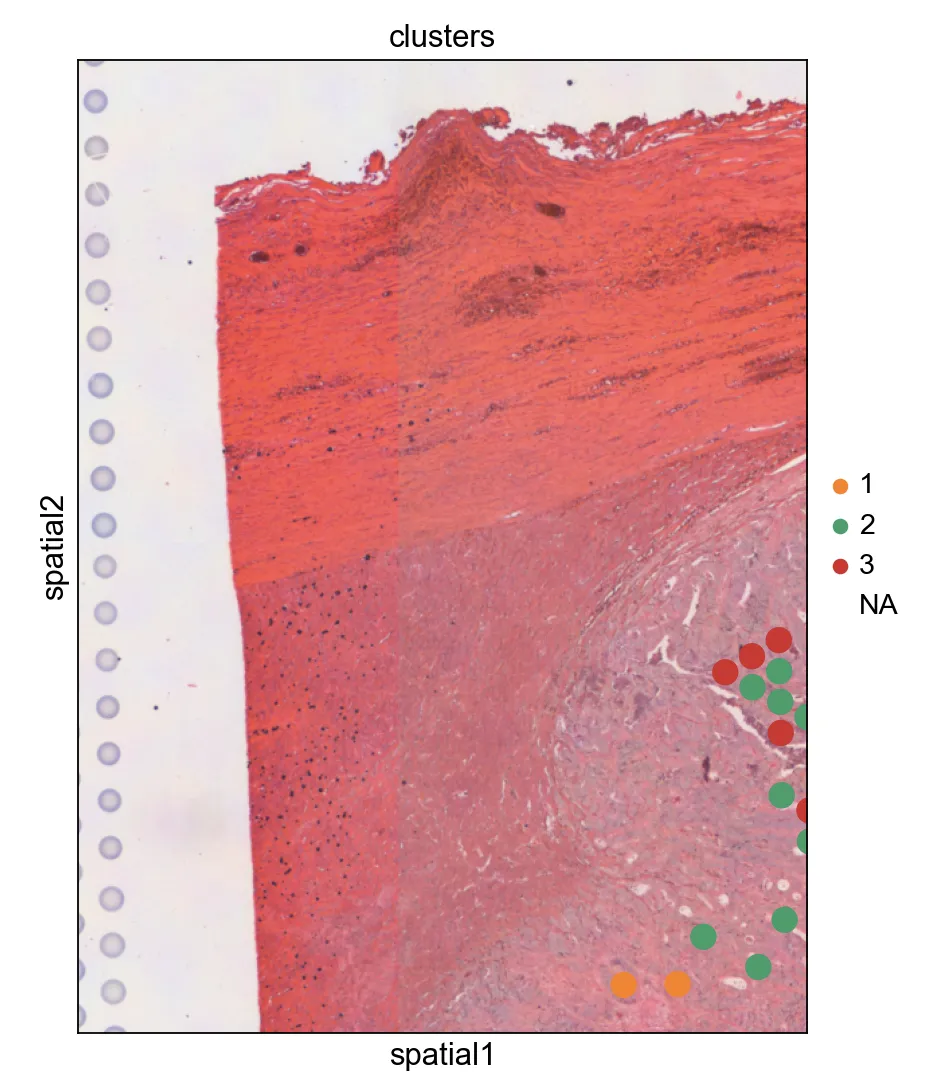
13.裁剪HE图片
adata.obsm['spatial'][:, 0].min(), adata.obsm['spatial'][:, 0].max()
# (np.int64(913), np.int64(4338))
adata.obsm['spatial'][:, 1].min(), adata.obsm['spatial'][:, 1].max()
# (np.int64(1751), np.int64(4177))
# [left, right, top, bottom]
sc.pl.spatial(adata, img_key="hires",
color="clusters", groups=["1","2","3"], size = 1.3,
crop_coord=[500, 2000, 1000, 3000])
随便剪裁了一下
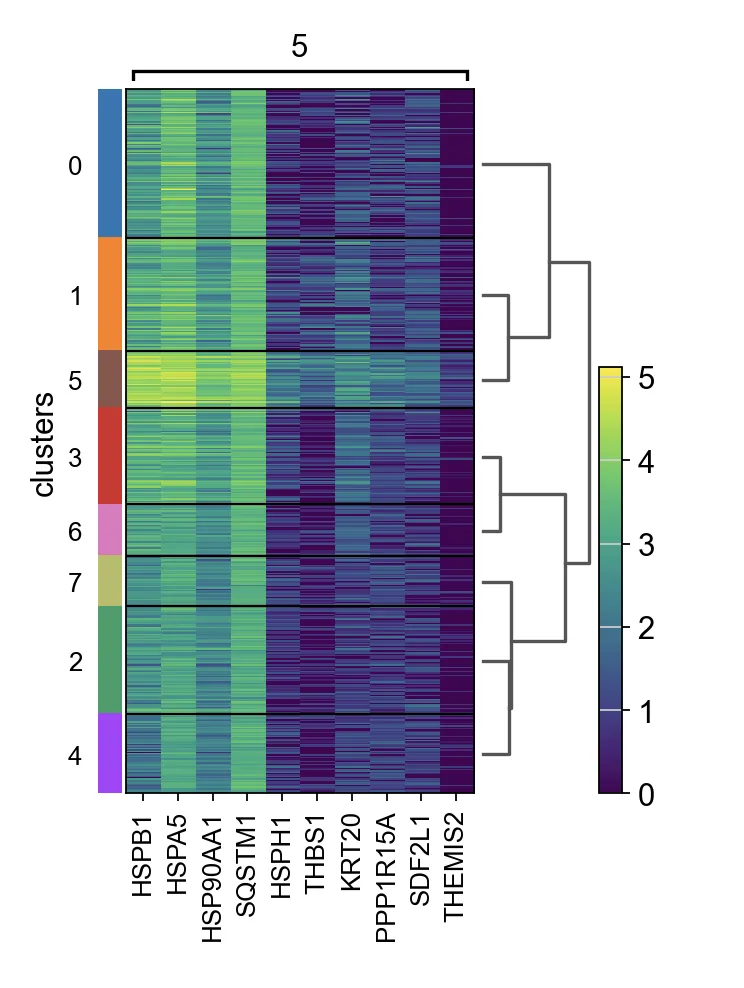
14.clusters差异基因分析并绘图
计算标记基因并绘制cluster5的差异基因在不同cluster中表达情况的热图。
sc.tl.rank_genes_groups(adata, "clusters", method="t-test")
sc.pl.rank_genes_groups_heatmap(adata, groups="5", n_genes=10, groupby="clusters")
15.cluster和基因表达展示
sc.pl.spatial(adata, img_key="hires", color=["clusters", "TP53"])
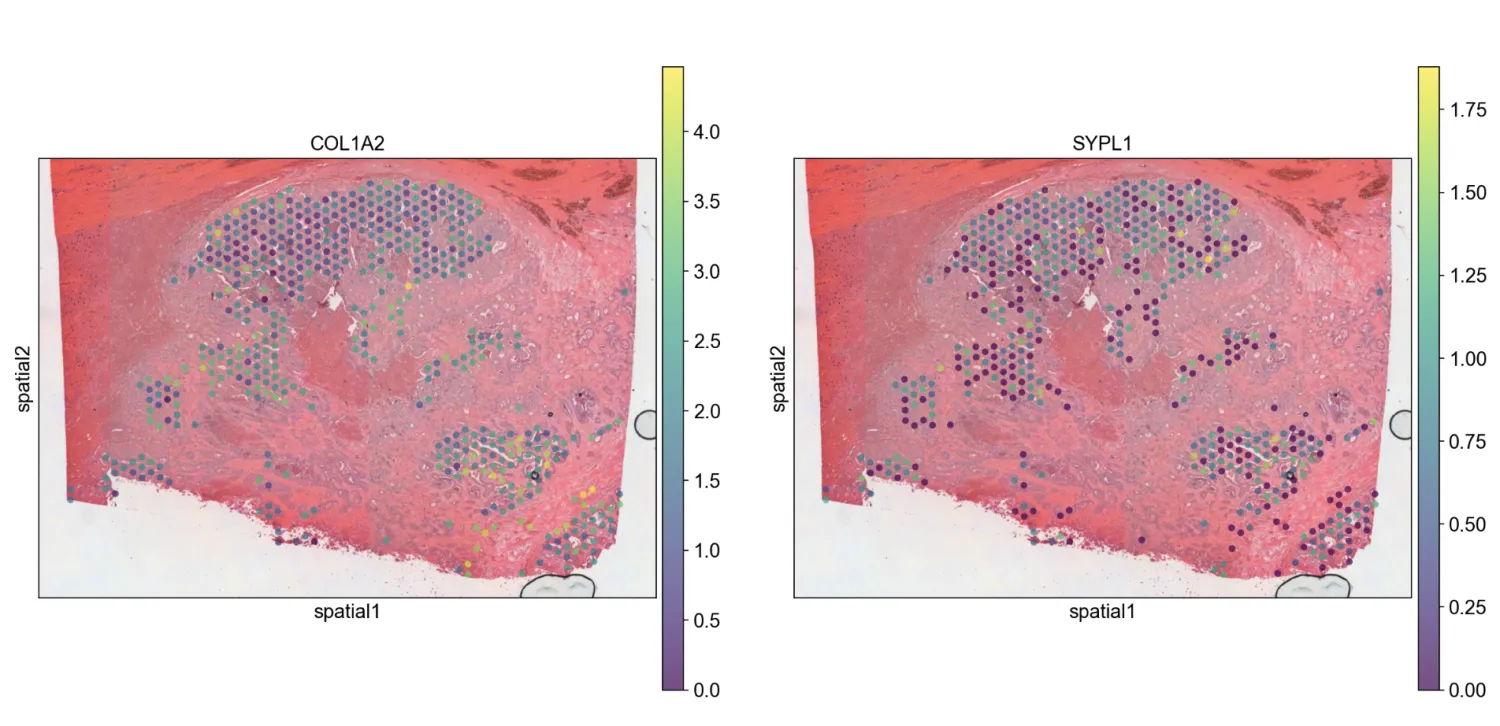
sc.pl.spatial(adata, img_key="hires", color=["COL1A2", "SYPL1"], alpha=0.7)

参考资料:
-
10XGENOMICS:https://www.10xgenomics.com/cn
-
scanpy空转:https://scanpy.readthedocs.io/en/stable/tutorials/spatial/basic-analysis.html
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -