UDP-鼠李糖合成酶基因的克隆与鉴定-文献精读76
何首乌中UDP-鼠李糖合成酶基因FmRHM1/2的克隆与鉴定

摘要
UDP-鼠李糖是一种由UDP-鼠李糖合酶(RHM)催化合成的鼠李糖供体,而鼠李糖是鼠李糖苷化合物的重要组成部分,植物中只有少数基因编码的酶参与UDP-鼠李糖生物合成。本研究基于何首乌(Fallopia multiflora(Thunb.)Harald.)转录组数据,首次克隆得到2个RHM基因(FmRHM1和FmRHM2),并进行生物学信息分析、体外功能鉴定及组织特异性分析。结果显示FmRHM1/2基因的开放阅读框均为2013 bp,均编码670个氨基酸,推测蛋白质分子质量均为75.6 kDa,理论等电点分别为6.20和7.19,具有RHM酶家族的特征信号序列(GxxGxxG/A和YxxxK);多序列比对与系统进化树显示,FmRHM与其他物种的RHM具有同源性。体外酶促反应结果显示,重组蛋白FmRHM1和FmRHM2均具有催化活性,可将UDP-葡萄糖转化为UDP-鼠李糖。组织特异性表达显示,FmRHM1和FmRHM2基因在根中的表达量最低,并与茎和叶相比均存在显著性差异。本研究首次报道了何首乌RHM,并验证了其催化功能,为进一步研究微生物合成UDP-鼠李糖奠定基础。
UDP-鼠李糖(UDP-Rha)是合成L-鼠李糖及苷的重要糖供体, L-鼠李糖是植物细胞壁果胶多聚糖RG-Ⅰ和RG-Ⅱ生物合成所必需的组分之一。植物中的鼠李糖基转移酶通过糖苷键将活化的鼠李糖与小分子连接起来[1], 构成的鼠李糖多糖参与植物细胞壁的形成, 在维管植物的生长及发育过程中发挥重要作用[2]; 细菌细胞表面多糖也含有鼠李糖, 对细胞生长和细菌间的相互作用起着重要作用[2]。UDP-Rha是黄酮类、皂甙类、三萜类和小酚类化合物等次生代谢物的糖苷类化合物合成的糖基供体[3]; 研究表明, 鼠李糖苷具有广泛的生物活性, 如抗炎作用[4]、抗病毒活性[5]、抗氧化作用[6]和抗癌作用[7]。自然界中至少存在两种形式核苷酸鼠李糖: dTDP-Rha和UDP-Rha[8, 9], dTDP-Rha存在于细菌中, 而UDP-Rha存在于真菌和植物中。在细菌中, 由dTDP-葡萄糖4, 6-脱水酶(RmlB)[10]、dTDP-4-酮-6-脱氧-D-葡萄糖3, 5-差向异构酶(RmlC)[11]和dTDP-4-酮-L-鼠李糖4-酮-还原酶(RmlD)[12]基因编码的3种酶连续催化dTDP-葡萄糖生成dTDP-Rha (图 1A)。真菌中则通过两种酶: UDP-葡萄糖4, 6-脱水酶(UG4, 6-Dh)和UDP-4-酮-6-脱氧-D-葡萄糖(UDP-4K6DG) 3, 5-差向异构酶-4-酮-还原酶通过两步催化合成UDP-Rha[13] (图 1B)。而植物中, 则存在一种同时具有UG4, 6-Dh、UDP-4K6DG 3, 5-差向异构酶和UDP-4-酮-L-鼠李糖(UDP-4KR)4-酮-还原酶活性的单酶, UDP-鼠李糖合成酶(RHM)整合了细菌dTDP-Rha生物合成途径中3种酶的功能, 直接催化底物UDP-α-D-葡萄糖(UDP-Glc)生成UDP-Rha (图 1C)。植物RHM由两个功能不同的结构域组成:分别是具有UG4, 6-Dh活性的N-末端和具有UDP-4K6DG 3, 5-差向异构酶和UDP-4KR 4-酮-还原酶活性的C-末端结构域[2]。

RHM是植物中控制鼠李糖苷合成途径中的关键节点酶, 将UDP-Glc转化为UDP-Rha, 并作为糖基供体, 用于鼠李糖分子类化合物的生物合成。目前已有化学合成和酶法合成UDP-Rha, 但在化学合成上无论是利用磷酸-鼠李糖与活化的核苷5'-单磷酸(NMP)进行偶联[14], 还是利用核苷二磷酸(NDP)与亲电子糖基供体反应, 在其末端磷酸上发生糖基化生成UDP-Rha[15], 都存在反应条件苛刻、反应效率和立体选择性低等问题, 实际应用价值有限。酶法合成是通过相关蛋白催化产物生成, 具有简单、快速、副产物少等优势, 已逐渐成为一种理想的替代化学合成的途径。目前, 已有报道利用拟南芥[2]、杨树[16]、玉米[17]等植物中的RHM酶, 以UDP-Glc为底物连续催化合成UDP-Rha的研究, 但在中药领域中却未见相关研究报道。
何首乌[Fallopia multiflora (Thunb.) Harald.]是我国著名的传统中药, 含有多种对人体有益的活性成分, 具有抗衰老、抗动脉粥样硬化、抗高血脂、抗肿瘤、抗炎、清除自由基、保肝等方面的生物活性[18]。何首乌中存在2, 3, 5, 4'-四羟基二苯乙烯-2-O-鼠李糖苷和槲皮素7-O-鼠李糖苷[19], 这暗示何首乌中存在RHM, 催化UDP-Rha的合成, 以提供鼠李糖糖基, 用于鼠李糖苷类化合物的生物合成。目前为止, 尚未有何首乌鼠李糖合成酶基因(FmRHM)的相关报道。本文通过何首乌转录组数据筛选, 首次克隆得到2条RHM基因(FmRHM1和FmRHM2)的全长cDNA序列, 并在体外进行原核表达获得重组蛋白; 通过以槲皮素、UDP-Glc、FmRHM基因和拟南芥AtUGT78D1编码的酶进行酶促反应, 检测槲皮素-3-O-鼠李糖苷的生成[16], 以鉴定FmRHM的体外功能。其次, 采用实时荧光定量PCR检测这两个基因在何首乌不同组织中的表达水平。何首乌中RHM基因功能的成功鉴定, 为合成生物学和代谢工程提供更多的植物基因用于微生物合成UDP-Rha, 为鼠李糖多糖及糖苷的生物合成奠定了基础。
材料与方法
材料
样品采自广东省德庆县大桥村, 经广东药科大学杨全教授鉴定为蓼科植物何首乌(F. multiflora)。取其根、茎、叶组织样品, 液氮速冻后置-80 ℃保存备用。用根、茎、叶提取总RNA, 检测FmRHM基因在不同组织中的特异性表达。所用的克隆、表达大肠杆菌(Escherichia coli) Transl-T1、Transetta (DE3)均购自全式金(TransGen Biotech, China)公司, 原核表达载体pET-28a (+)由实验室传代冻存。引物合成及片段测序送北京擎科新业生物技术有限公司(Tsingke, China)进行。拟南芥AtUGT78D1基因在GenBank的登录号为AY056312.1, 序列送苏州泓迅生物科技有限公司合成。槲皮素、槲皮素3-O-葡萄糖苷、槲皮素3-O-鼠李糖苷均购自成都瑞芬思生物科技有限公司。
何首乌总RNA的提取与cDNA的合成
按照RNAprep Pure试剂盒(Tiangen, China)操作说明提取何首乌总RNA, 利用Nano-100检测RNA浓度和纯度, 同时利用1.0%琼脂糖凝胶电泳检测RNA的完整性。使用反转录试剂盒(TaKaRa, Japan)将总RNA反转录为第一链(cDNA), -20 ℃保存备用。
FmRHM基因序列全长克隆
根据从何首乌转录组数据中筛选的2条FmRHM基因序列, 设计特异性扩增引物, 设计原理基于pEASY-Uni Seamless Cloning and Assembly Kit (TransGen Biotech, China)说明书, 分别在一对引物的5'端引入一段和线性化载体(pET28a)两端相同的序列(引物序列见表 1)。利用扩增试剂盒(TOYOBO, Japan), 以何首乌RNA的反转录产物为模板, 进行聚合酶链式反应(PCR)扩增FmRHM基因片段。PCR反应体系为: cDNA 1 μL, KOD-Plus-Neo (1 U·μL-1) 1 μL, 10×PCR Buffer for KOD-Plus-Neo 5 μL, 2 mmol·L-1 dNTPs 5 μL, 25 mmol·L-1 MgSO4 3 μL, 10 μmol·L-1上下游引物各1.5 μL, ddH2O补足至50 μL。反应程序: 94 ℃预变性2 min; 94 ℃变性30 s, 58 ℃退火30 s, 72 ℃延伸1 min, 35个循环后; 72 ℃延伸10 min; 4 ℃保存。1.0%琼脂糖凝胶电泳检测PCR产物, 利用TaKaRa胶回收试剂盒回收PCR产物。使用无缝克隆试剂盒(TransGen Biotech, China), 将PCR产物与BamH Ⅰ酶切后的pET28a载体连接。转化到Transl-T1菌株中, 在含卡那霉素抗性的平板上进行筛选, 并经过菌落PCR检测后送擎科公司测序。

何首乌FmRHM1和FmRHM2的生物信息学分析
利用ExPASy Proteomics Server在线软件Protparam对2个FmRHM基因编码蛋白的氨基酸组成、蛋白质分子质量、理论等电点及稳定性等理化性质进行分析; 通过SOPMA预测蛋白质二级结构; 利用SWISS-MODEL在线软件构建FmRHM1和FmRHM2蛋白的三级结构模型; 在线软件TMHMM 2.0进行蛋白质跨膜结构分析; 将所获得的FmRHM编码的氨基酸序列在NCBI中进行蛋白Blast比对分析, 通过DNAMAN软件与其他物种的RHM基因编码的氨基酸序列进行同源性分析; 通过MEGA 6.0软件构建Neighbor-joining系统进化树, 进化距离的计算采用泊松校正法, Bootstrap重复次数为1 000次。
FmRHM蛋白原核表达
挑选测序验证后的转化子, 过夜培养, 提取pET28a-FmRHM重组质粒, 转化Transetta (DE3)感受态大肠杆菌中培养。挑取阳性克隆进行PCR检测, 并送测序验证表达系统正确性。挑选序列正确菌株, 按1:100比例加入到适量含有50 μg·mL-1卡那霉素的LB液体培养基中; 37 ℃、250 r·min-1振荡培养至OD600达0.6~1.0左右; 加IPTG至终浓度0.4 mmol·L-1, 30 ℃、200 r·min-1培养细胞5 h, 诱导产生重组FmRHM蛋白。经诱导表达的培养物, 以4 000 ×g离心15 min收集菌体, 用ddH2O清洗菌体2次后, 菌体悬浮于缓冲液(50 mmol·L-1 Tris-HCl, 1 mmol·L-1 EDTA, 10%甘油, 1 mmol·L-1 PMSF)中, 超声破碎(30%功率, 超声5 s, 间歇5 s, 工作5 min)。细胞破碎液于4 ℃、13 000 r·min-1离心15 min, 去除细胞碎片。取粗酶液加入6×loading buffer混匀, 沸水浴5 min, 12 000 ×g离心5 min, 上样10 μL进行10% SDS-PAGE电泳, 电泳结束后将胶置于考马斯亮蓝染色1 h, 用脱色液进行背景脱色, 检测蛋白表达情况。
拟南芥AtUGT78D1表达载体构建及体外表达
将AtUGT78D1连接到BamH Ⅰ和Not Ⅰ酶切的pET28a载体上, 构建重组表达载体。克隆菌株扩大培养, 提取重组质粒, 转化到Transetta (DE3)中培养, 挑选阳性克隆菌, 按上述项下的操作进行AtUGT78D1重组蛋白的体外原核表达及蛋白提取, 并进行10% SDS-PAGE电泳检测。
酶促反应检测
酶促反应体系:总体积为400 μL, 含有0.1 mmol·L-1槲皮素、0.5 mmol·L-1 UDP-Glc、0.5 mmol·L-1 NADPH、0.5 mmol·L-1 NAD+、AtUGT78D1和FmRHM粗酶液各193 μL。以未连接目的基因的pET28a空载表达菌在同等表达条件下的蛋白粗提物进行酶促反应作为阴性对照。混匀后于30 ℃下孵育12 h, 加入等体积甲醇终止反应。13 000 r·min-1离心30 min, 取上清过0.22 μm滤膜, 进行HPLC检测。检测条件如下: Waters 1525高效液相色谱仪系统; 色谱柱: Phenomenex 00G-4252-E0 (4.6 mm×250 mm, 0.45 μm); 流动相:甲醇-0.1%磷酸溶液(55:45, v/v); 流速: 1 mL·min-1; 进样体积: 10 μL, 30 ℃柱温, 366 nm的波长下进行检测。
何首乌FmRHM基因的组织特异性表达分析
利用qRT-PCR方法检测何首乌FmRHM基因在不同组织中的表达水平。使用TaKaRa的反转录试剂盒(PrimeScriptTM RT reagent Kit with gDNA Eraser)进行反转录生成单链cDNA。使用TB GreenTM Premix Ex TaqTM Ⅱ试剂盒(TaKaRa, Japan), 在CFX96 Touch (BioRad, USA)仪上进行扩增。选取何首乌Actin基因作为目标基因定量表达的内参基因, 利用Integrated DNA technologies在线软件设计引物, 引物序列见表 1。每个样品设3个重复, 重复3次。扩增体系中含有10 μL TB Green Premix Ex Taq Ⅱ (2×)、上下游引物(10 μmol·L-1)各0.8 μL、模板2 μL, ddH2O补至总体积为20 μL。反应程序: 95 ℃预变性30 s; 95 ℃变性5 s, 60 ℃退火/延伸30 s, 40个循环后; 95 ℃变性10 s, 65~95 ℃做熔解曲线分析, 每个温度以每步0.5 ℃上升, 每个温度停留5 s。根据熔解曲线判断RT-PCR产物的特异性, 相对定量分析采用2-ΔΔCt方法进行, 结果采用GraphPad Prism 7.0进行各组间方差分析。
结果与分析
1 何首乌FmRHM基因全长cDNA克隆
以何首乌cDNA为模板进行扩增, 通过PCR扩增后, 2条基因的PCR产物均约为2 000 bp, 与预期大小相近, 如图 2所示。将纯化后的PCR产物连接到pET28a载体上, 进行测序。测序结果显示克隆的片段与转录组数据中的基因序列一致, 其序列长度均为2 013 bp, 均编码670个氨基酸, 基因命名为FmRHM1和FmRHM2。

2 FmRHM1和FmRHM2生物信息学分析
2.1 FmRHM1和FmRHM2理化性质分析、亚细胞定位、跨膜区域分析
通过Protparam软件预测重组蛋白的理化性质(表 2), 推测的FmRHM1和FmRHM2编码的蛋白均属于稳定和亲水性蛋白; 且TMHMM 2.0预测显示何首乌FmRHM1和FmRHM2蛋白均无跨膜区域。

2.2 FmRHM1和FmRHM2蛋白的二级结构分析及三维结构预测
结果显示, 预测FmRHM1和FmRHM2基因编码蛋白的二级结构由无规则卷曲(random coil)、α-螺旋(α-helices)、延伸链(extended strand)和β-折叠(β-turn)组成(图 3), 且大部分由无规则卷曲和α-螺旋组成(表 3), 推测两者是其主要的二级结构元件, 同时α-螺旋、β-折叠和延伸链散布于整个蛋白中。


将FmRHM1和FmRHM2的氨基酸序列通过SWISSMODEL Workspace在线分析软件建立三维结构模型, 选择AtRHM (PDB No.: 4QQR)的蛋白结构为模板[20], 对FmRHM1和FmRHM2结构进行预测, 所得三维结构如图 4所示。两个蛋白与AtRHM蛋白的相似度分别为82%和83%, 因FmRHM1和FmRHM2的氨基酸序列相似度很高, 所以在蛋白三维结构模型上较难看出区别。

2.3 何首乌FmRHM氨基酸序列和系统进化树分析
将何首乌FmRHM氨基酸序列与GenBank蛋白数据库中进行同源比对, 通过DNAMAN软件与多种植物进行多序列比对分析, FmRHM1和FmRHM2与其他物种RHM同源性很高, 达85%以上。将FmRHM1和FmRHM2与拟南芥、虎眼万年青、茶树的RHM进行详细比对, 发现FmRHM与这些植物RHM的N-端和C-端高度相似, 而这两个功能单元, 在拟南芥RHM2的体外酶活性分析发现, N-端区具有较强的UDP-葡萄糖4, 6-脱水酶活性, 而C-端区域则同时具有UDP-4K6DG 3, 5-差向异构酶和UDP-4KR 4-酮-还原酶活性[21]。且FmRHM1和FmRHM2每个功能单元中均存在NADP(H)结合位点(GxxGxxG/A)和类似RmlD酶结构的活性中心, 即YxxxK模序(图 5)。

为了进一步了解何首乌FmRHM1/2蛋白在植物RHM家族中的进化位置, 将FmRHM1/2与NCBI中51条RHM氨基酸序列进行系统发育分析, 其中包括细菌、真菌、蕨类植物和苔藓门, 以及植物中RHM序列, 包括乔木类植物甜橙、葡萄等, 模式植物拟南芥, 以及其他植物如大豆、虎眼万年青等, 利用MEGA 6.0构建系统进化树。结果表明, 不同来源的RHM最后聚为一支, 表明其具有共同的祖先, 且在被子植物中分为双子叶植物和单子叶植物两类, 何首乌中的2个RHM均归属于双子叶植物一类; 另外, FmRHM2与双子叶植物陆地棉距离较近, 而FmRHM2则单独聚为一支(图 6)。

3 FmRHM与AtUGT78D1原核表达分析
本研究用pET28a为表达载体, 利用同源重组原理构建pET28a-FmRHM重组原核表达载体, 并转化Transetta (DE3)感受态大肠杆菌进行诱导表达蛋白。SDS-PAGE电泳结果表明, 经诱导后, 与未连接目的基因的pET28a空载诱导蛋白及未诱导的含目的基因载体表达的蛋白对比。在约70 kDa和52 kDa附近, 诱导样品6、7和8出现明显的蛋白条带(图 7)。FmRHM1和FmRHM2的预测分子质量大小均约为75 kDa, AtUGT78D1的预测分子质量约为50 kDa, 所以这3个蛋白条带分别为诱导表达的FmRHM重组蛋白和AtUGT78D1重组蛋白。

4 FmRHM酶促反应检测
在酶促反应液中加入UDP-鼠李糖合成酶和糖基转移酶共同反应, 检测反应液中糖苷的形成。HPLC检测结果显示(图 8), 所有样品均在保留时间约为4.17 min处出现色谱峰, 与标准品中的槲皮素-3-O-葡萄糖苷(S1)的出峰时间相近, 说明反应液中均生成槲皮素-3-O-葡萄糖苷; 在槲皮素(S3)的出峰时间8.48 min处未见明显峰, 说明槲皮素大部分已被AtUGT78D1酶转化为糖苷, 所以检测量较低; 更重要的是, 重组表达质粒组与空载对照组相比, FmRHM1和FmRHM2组均在保留时间约为5.10 min处出现明显色谱峰, 与槲皮素-3-O-鼠李糖苷(S2)的出峰时间一致, 对照组却未在此出现明显峰, 证明重组质粒组的反应液中生成了UDP-Rha, 从而使AtUGT78D1酶能利用除UDP-Glc外的UDP-Rha为糖基供体, 生成槲皮素3-O-鼠李糖苷。结果验证了所得FmRHM重组蛋白均具有生物学活性, 可在体外催化UDP-Glc转化生成UDP-Rha。

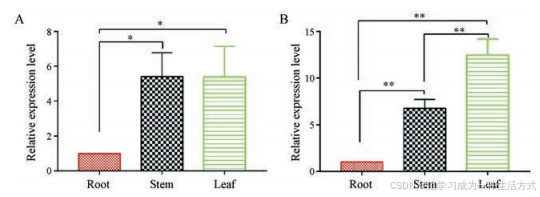
5 FmRHM基因组织特异性表达分析
采用qRT-PCR方法分析FmRHM1和FmRHM2基因在何首乌不同组织(包括根、茎、叶)中的相对表达量, 图 9表示两个基因在根、茎、叶中的相对表达水平, FmRHM1和FmRHM2在根中的表达量均最低。尽管FmRHM1和FmRHM2编码的蛋白在体外具有相同功能, 但它们的表达水平是不同的。显著性差异分析结果显示, FmRHM1在茎、叶中的表达水平与根之间的差异均具有显著性, 但茎叶间的相对表达量相近, 差异不显著; 而FmRHM2在不同组织间的表达水平均存在显著性差异, 表达量呈现叶 > 茎 > 根的结果。
讨论
鼠李糖是许多天然有效产物的一部分, UDP-Rha是一种活化形式的L-鼠李糖, 参与鼠李糖苷的生物合成, RHM是植物鼠李糖合成的关键酶。目前, 尚未有何首乌中关于RHM基因的相关研究, 本研究首次在从何首乌中扩增得到2个RHM基因(FmRHM1和FmRHM2)全长, 并对其进行生物信息学分析; 以拟南芥中RHM蛋白为模板进行同源建模, 构建了FmRHM1和FmRHM2蛋白的三维结构; 多序列比对分析发现2个FmRHM含有植物中RHM的2个高度保守结构(GxxGxxG/A和YxxxK), GxxGxxG/A是酶促反应辅因子NADP(H)的结合模序, 而辅因子NAD+则结合YxxxK结构, 有趣的是在序列的N-端和C-端均存在这两种结构, 暗示所得的两个FmRHM均具有将UDP-Glc转化为UDP-Rha的催化能力; 进化分析结果表明FmRHM与其他物种RHM存在同源性。另外, 本研究通过构建原核表达载体pET28a-FmRHM, 诱导表达重组蛋白, 并通过加入FmRHM和拟南芥糖基转移酶进行酶促反应检测鼠李糖苷的生成, 证实了表达的FmRHM蛋白具有催化合成UDP-Rha的功能。植物中含糖类化合物的生物合成存在许多未知, 因此, 采用qRT-PCR方法研究FmRHM基因在何首乌不同组织中的表达水平, 旨在探讨RHM在鼠李糖类小分子化合物生物合成中可能存在的作用。数据显示何首乌RHM基因存在组织特异性表达, 且FmRHM1和FmRHM2在不同组织中的表达水平不同, 总体来说, 在叶和茎中均表现出较高的表达量, 暗示RHM基因可能参与某种鼠李糖类化合物的合成, 在叶和茎发育过程中起主要作用。
目前, 通过生物合成核苷酸糖的实际应用还很少, 并且合成方法还不够完善, 如UDP-Xyl[22]合成过程中存在需要多种酶参与, 反应步骤多, 反应效率较难控制, 合成费用高等问题。然而, 在本研究中所提出的何首乌基因所编码的酶, 只需要单酶参与反应便能合成UDP-Rha。作为鼠李糖基供体, UDP-Rha是鼠李糖苷生物合成所必需的, 也是植物细胞壁组成中不可缺少的。毫无疑问, 这两个基因的成功鉴定为全面分析含鼠李糖多糖的生物合成途径奠定了良好的基础, 提供了一种简单快速合成UDP-鼠李糖的方法, 目前该技术尚未在商业上使用。本研究阐述了利用植物基因合成小分子糖苷的方法, 为天然产物鼠李糖苷的生物合成奠定基础。