板块模型构建、k点选定及Miller指数对表面分类
1表面的重要性
表面对于催化、界面科学、电子制造,半导体制造等很多领域都是非常重要的。理解表面的几何构型和电子结构十分有意义。在一些科研项目中,表面科学实验和DFT计算成功的结合在一起。DFT计算常常和超高真空表面科学的实验技术相结合,例如:扫描隧道显微镜()、程序升温脱附(
)、X射线光电子能谱(
),从而可以确定金属、金属氧化物、纳米颗粒、碳化物和硫化物的表面结构。例如,1959年通过对
表面进行电子衍射实验,表明该表面具有复杂对称性,并且形成表面的原子排列与体相
晶体结构完全不同。由于
在微电子技术的重要性,详细了解其表面结构是十分重要的。知道1992年才完成一个很大的超晶胞的DFT计算,从而对
表面重构进行了有效性测试,并且该测试的认可度很高。这个计算不仅明确给出了该表面每个原子的位置,并且可以用于模拟该表面的STM成像,有助于解释实验所得图像。
下面将介绍如何用DFT计算考察固体表面。
2周期性边界条件和板块模型
当研究的对象是一个表面时,理想的模型应该是两个方向上无限大,在沿表面法线上有限大的一条长材料。为了在建模上实现这一点,可以在两个方向上应用周期性边界条件优势,在法线方向上不可以。虽然已经有相关计算软件能够实现这一点,但在研究表面的时候,更加普遍使用的计算程序
是在三个坐标方向上都应用了周期性边界条件。基本概念如下图所示,在超晶胞的竖直方向上。只有一部分区域含有原子。在超晶胞下部,这些原子在x和y方向上充满了整个超晶胞,但是在这些原子的上方,也就是超晶胞的顶部区域是没有原子的,因此这种模型称之为板块()模型。

当该超晶胞在所有三个方向上都重复时,就定义了一系列堆垛在一起的,并由空白区域所分隔开的固体材料板块。如下图所示。在z方向上分隔开周期性板块图像的空白区域称之为真空区()。当使用这种模型块时一定需要足够大的真空区,以便材料的电荷密度在真空区逐渐消失为零,从而使板块的顶面与相邻板块的底面之间不存在作用。

下图所示就是在真空区内所观察使用这种方法所定义的原子视图,可以看到:超晶胞实质上定义了两个表面,一个是上表面,一个是下表面。

假设对金属(例如
)的表面进行计算,应当如何建立上述的板块模型?使用超晶胞的晶格矢量分别和笛卡尔坐标系的
轴一致,其中
轴和超晶胞表面法线方向一致。对于fcc金属,其晶格常数与常规晶胞中立方体边长相等。超晶胞的矢量应该是:
式中:为晶格常数。
图4.1中原子的分数坐标是:
如上所述,定义了含有5层独立原子的板块,且在z方向上,两层原子间距为,真空区的宽度为
。对于该超晶胞,板块的每层原子均含有两个独立原子。在定义
时,数值5有一点随意。但可以精确的是,该矢量必须做够大,从而保证该超晶胞具有足够的真空区;但真空区越大,则所需的计算量越大。在实际计算过程中,需要对若干个具有不同真空区宽度的模型,检验真空区的电荷密度,从而判断需要多少真空区才是对这个板块而言足够大的。理想情况是真空区边缘的电荷密度接近于0。如果读者想要做该测试,需要注意的是:在改变
长度的同时,原子分数坐标的
值也需要相应的调整,以保证材料中相邻两层原子之间是正确的物理间距。
(分数坐标与笛卡尔坐标
分数坐标是把点阵矢量看成是单位矢量下确定原子位置; 笛卡尔坐标是绝对坐标,是直角坐标系与斜角坐标系的统称,考虑点阵常数的大小。
Basically, the fractional coordinates show the fraction of each unit cell vector that contributes to the position of that atom。根本上来说,分数坐标给出了每个原子在元胞的相对位置
晶格参数:
A (Ax Ay Az) 2.46 0.00 0.00
B (Bx By Bz) -1.23 2.13 0.00
C (Cx Cy Cz) 0.00 0.00 10.00
一个点的分数坐标 (a, b, c) (0.667 0.333 0)
x = Axa+Bxb+Cxc = 2.46*0.667-1.23*0.333+0*0=1.23
y = Aya+Byb+Cyc = 0*0.667+2.13*0.333+0*0=0.71
z = Aza+Bzb+Czc = 0*0.667+0*0.333+10*0=0
这个点的笛卡尔坐标 (x, y, z) (1.23 0.71 0)
所以,笛卡尔坐标并不是原子在我们作为参考的直角坐标系中的绝对位置,而是在元胞基矢构成坐标系中,原子的绝对位置,而分数坐标,就是原子在元胞基矢构成坐标系中原子的相对位置。对于一个斜角坐标系,显然原子的相对位置比较好判断,所以,我们一般在POSCAR中用到的是其分数坐标,如果需要用到笛卡尔坐标,就用上面的公式转换。)
上述讨论中的板块模型包括有金属的5层原子。当然,一个真实表面时一块材料的最边缘部分,且除非在很特殊的情况下,一班材料都至少几微米厚,与上述5层表面模型完全不同。这就引出了一个问题:多少层原子才是具有现实意义的?通常而言,原子层数越多越好,但所用的原子层数越多,那么计算时间一定会延长。关于怎么判断原子层数已经达到足够收敛了,这一点取决于所研究材料的物质本性和所需计算的参数。如同真空区宽度一样,这个问题也可以通过如下方法找到答案:计算并观察某些特性(如表面能和吸附能)随着原子层数逇增加时如何变化的。在选定模型总的原子层数时,通常是在计算成本和物理精度之间进行折中。
3选定表面计算的 点
点
对于板块计算,可以采用方法选定
点。为了进一步直观的了解这一点,可以计算一下与上述超晶胞相关的倒易晶格矢量。可得
,
。因为超晶胞在实空间中晶格矢量越长,对应着在倒易空间越短。这表明在倒易空间中求解积分的近似解时,在
方向上并不需要那么多的k点。对于该超晶胞,使用一个
的
点网格。其中,对于该几何构型而言,
是较为合适的。
超晶胞z方向上较大的尺寸包含了真空区,这一事实引入了另一个有关点的选择经验。如果真空区足够大,则在距离板块边缘很短的位置上,电荷密度就消失为0。这意味着:在
方向上只使用一个
点就能得到精确的结果。因此,对于板块计算常常使用
的
点网格。其中,所选定的
和
能够在表面模型的晶面内,对其
空间进行足够精确的取样。
4采用 指数对表面分类
指数对表面分类
对于图4.3所示表面,可以想象成将体相晶体劈裂()后所形成的。真实的晶体劈裂,可以沿着不同晶面对晶体进行劈裂,形成不同晶体排列的表面。这意味着需要采用一种标注方法,用于定义所讨论的表面。
图4.4给出了表面的晶面与体相晶体结构的对应关系。该晶面的朝向可以用与与该晶面相垂直的法相矢量来描述。从下图可以看出,这个矢量的一个有效选择就是
,也可以选择
等相平行的矢量。
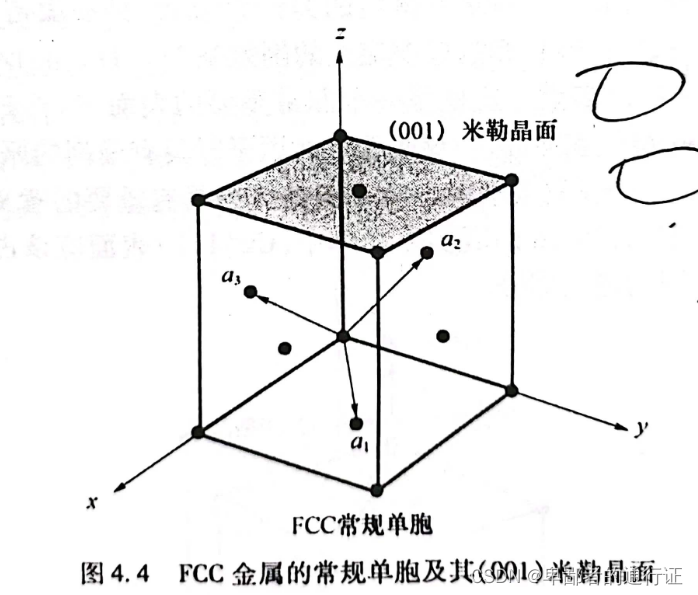
为了确定应该使用这些矢量中的哪一个,通常需要先确定该晶面与材料原胞或者常规单胞
三轴的截交点。将这些截交点的倒数同乘以某个因子,从而将每个倒数都化成整数,并使每个倒数都尽可能小,最终所得的数值就称为该表面的
指数。如上图所示,该晶面与常规单晶胞的
轴相交于1(以晶格常数为单位),但是和
轴并不相交,则这些截交点的倒数是(0,0,1),该表面可以表示成
。
如果定义该表面垂直方向的矢量需要加个负号,则其指数的相应分量上也要加个横杠。但对于这种晶体结构来说,由于对称性,不严格区分。

图4.5显示材料的另一种表面, 该表面为。这也是一个非常重要的表面,对于
材料的所有
指数表面而言,该表面的原子层具有较高的原子密度。也正是如此,这个表面在平衡态的真实晶体中具有重要的意义。上述讨论表明:在
的真实多晶体中,
表面应该占据了该晶体整个表面的绝大部分。
图4.6显示了表面,与z轴不相交。

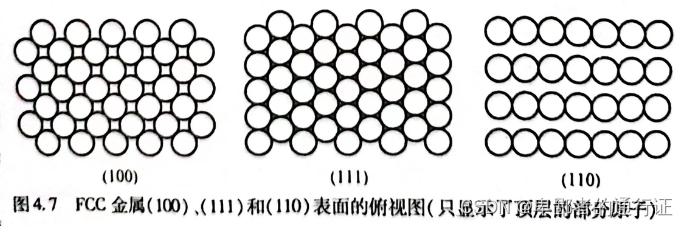
图4.7显示了表面的俯视图。这些试图给出了每个表面的不同对称性。这三种
表面全是原子尺度的平直表面,即:任意种表面上的每个原子都具有相同的配位关系,且在表面法向上的坐标也相同。这些表面综合在一起称之为
材料的低指数表面。其他晶体结构也具有自身的低指数表面,但与
结构不同的是:其他结构的低指数表面往往具有不同的
指数。例如,对于
材料,其原子密度最高的表面是
表面。
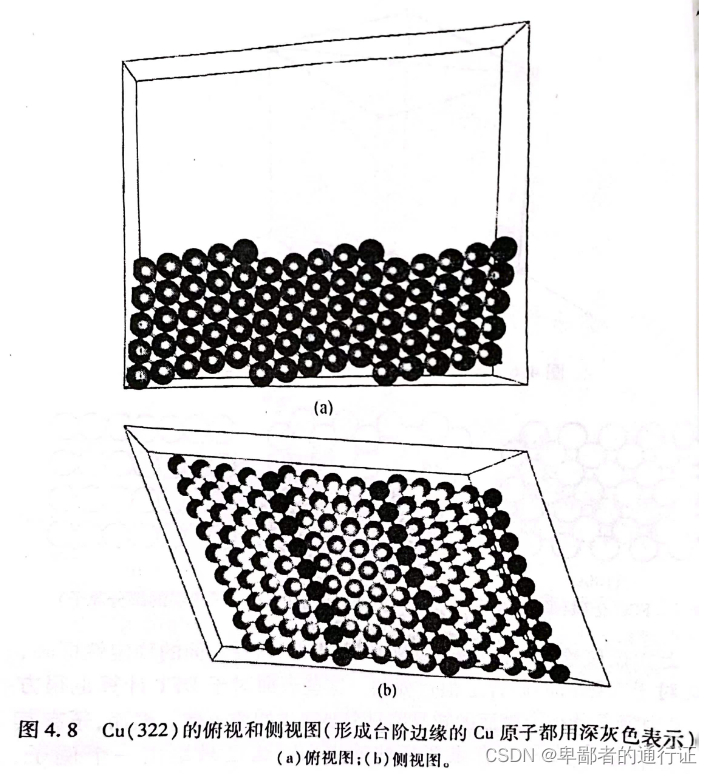
上文仅仅给出了低指数表面的一些示例这些表面的稳定性高,对于实际研究很重要。这些表面的计算也很方便,因为定义低指数表面的超晶胞只需要适当的原子数。图4.8是
表面的俯视图和侧视图。该表面许多局部区域看起来很像
表面,并且这些区域均由一个原子高度的台阶所分隔开来。相比于表面上的其他原子,这些位于台阶边缘处的原子具有较低的配位数,这往往导致具有更高的反应活性。在催化合成氨时,台阶边缘的反应活性原子就扮演着至关重要的作用。尽管
要比低指数表面更为复杂,但是该表面的晶面任然具有周期性,因此任然可以使用超晶胞来描述该表面。