万字长文 - Nature 综述系列 - 给生物学家的机器学习指南 2 (传统机器学习方法如何选择)...
万字长文 - Nature 综述系列 - 给生物学家的机器学习指南 1

传统的机器学习
我们现在讨论几种关键的机器学习方法的优势和劣势。表1显示了不同机器学习方法的比较。我们首先讨论不基于神经网络的方法,有时被称为“传统机器学习”。
图3显示了一些传统的机器学习方法。可以使用各种软件包来训练这些模型,包括
Python中的scikit-learn (『SKLearn』最全应用指南 (万字长文) | 图解机器学习实战)、
R中的caret包(Classification and Regression Training)(基于Caret和RandomForest包进行随机森林分析的一般步骤 (1))
和Julia中的MLJ (Machine Learning in Julia)。
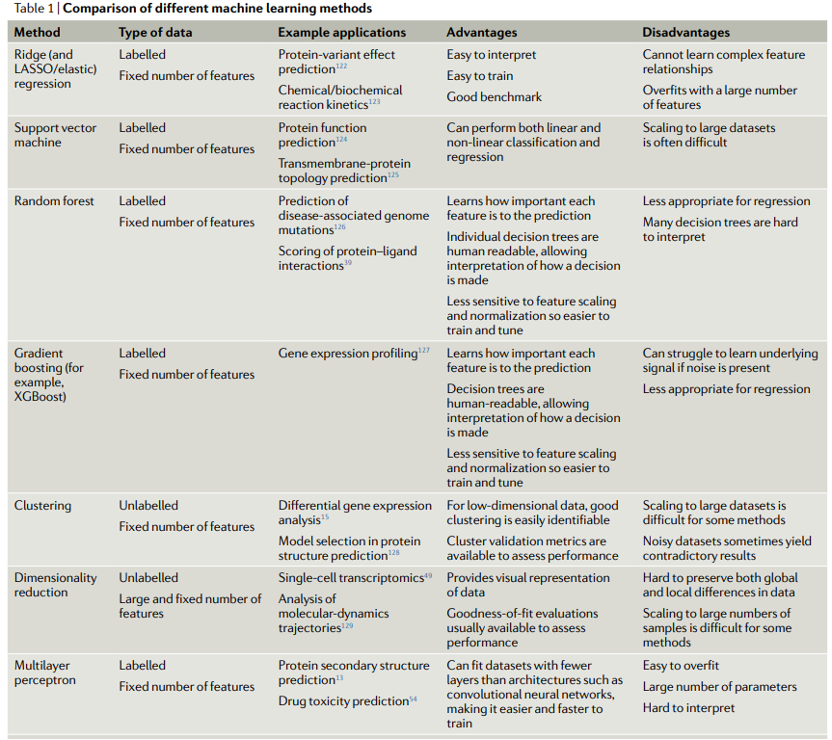
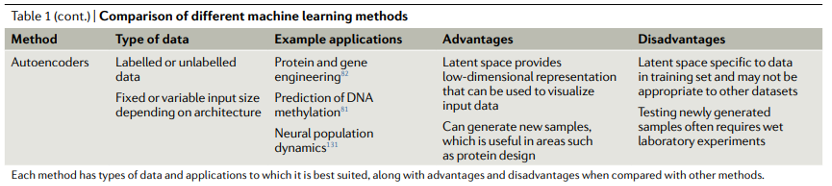

图3 | 传统机器学习方法。a | 回归分析找出因变量(观察属性)与一个或多个自变量(特征)之间的关系;例如,根据一个人的身高预测其体重。b | 支持向量机(SVM)对原始输入数据进行变换,以便在变换后的版本(称为“隐变量”)中,属于不同类别的数据之间有一个尽可能宽的明确间隙。在这个例子中,我们展示了如何预测蛋白质是有序还是无序,其中轴表示变换数据的维度。c | 梯度提升使用一组弱预测模型(通常是决策树)进行预测。例如,可以根据分子描述符(如分子量和特定化学基团的存在)预测活性药物。单个预测器以分阶段的方式组合,以得出最终预测。d | 主成分分析(PCA)找到一系列特征组合,这些特征组合可以在互相正交的同时最好地描述数据。它通常用于降维。在描述一个人的身高和体重的情况下,第一个主成分(PC1),相当于身高和体重的线性组合,描述了强烈的正相关,而PC2可能描述了与这些变量没有强相关的其他变量,例如体脂百分比或肌肉质量。e | 聚类:使用算法对相似对象的集合进行分组(例如,根据基因表达谱对细胞类型进行分组)。
线性回归:一种模型,假设输出结果可以通过输入特征的线性组合计算得出;即,每个输入特征乘以一个单一参数,然后将这些值相加。这些模型的预测方式很容易解释。
核函数:应用于每个数据点的转换,将原始点映射到一个相对于其类别可分离的空间中。
非线性回归:一种模型,其中输出是通过输入的非线性组合计算得出的;即,在预测过程中,可以通过诸如乘法等操作组合输入特征。这些模型可以描述比线性回归更复杂的现象。
k近邻算法:这是一种分类方法,其中根据训练集中k个最相似点的已知(基准真相)类别通过多数投票规则对数据点进行分类。k是一个可以调整的参数。通过对k个最近邻居的属性值求平均,也可以用于回归分析。
当开发用于生物数据的机器学习方法时,通常应先在传统的机器学习算法视中寻找最适合给定任务的方法。深度学习可以是一种强大的工具,目前无可否认是一种潮流。然而,它仍然被限制在它所擅长的应用领域:当存在大量可用数据时(例如,数百万数据点);当每个数据点具有许多特征时;以及当特征高度结构化时(特征彼此之间具有清晰的关系,例如图像中的相邻像素)。DNA、RNA和蛋白质序列以及显微图像等数据是可以满足这些要求并有应用深度学习的成果案例。然而,即使满足了其他两个要求,对大量数据的需求会使深度学习成为一个糟糕的选择。
与深度学习相比,传统方法开发和测试给定问题的速度要快得多。与支持向量机(SVM)和随机森林等传统模型相比,开发深度神经网络的架构,然后对其进行训练,可能是一项耗时且计算昂贵的任务。尽管存在一些方法,但对于深度神经网络,估计特征的重要性(即,每个特征对预测的贡献有多重要)或模型预测的置信度仍然是一个难点,而这两者对生物研究通常是必不可少的。即使深度学习在技术上对于特定的生物预测任务是可行的,如果可能的话,训练一种传统的方法来与基于神经网络的模型进行比较仍然是明智的。
传统方法通常期望数据集中的每个示例具有相同数量的特征,而这并不总是可能的。一个明显的生物学例子是,当使用蛋白质、RNA或DNA序列时,每个序列都有不同的长度。要对这些数据使用传统方法,可以使用填充和滑窗等简单技术来更改数据,使其特征长度相同。“填充”意味着对每个示例进行填充,并添加诸如零等的值,直到其大小与数据集中最大的示例相同。相比之下,滑窗将单个实例的特征缩短到给定大小(例如,仅使用长度在100以上的蛋白质序列数据集中每个蛋白质的前100个氨基酸)。
分类和回归模型的使用。对于图3a中所示的回归问题,岭回归(ridge regression, 带有正则化项的线性回归)通常是开发模型的很好的起点,因为它可以为给定任务提供快速且易于理解的基准结果。线性回归的其他变体,如LASSO回归和弹性网络回归,在需要模型依赖可用数据中的最小数量的特征时也值得考虑。不幸的是,数据中特征之间的关系通常是非线性的,因此对于这些情况,使用诸如SVM之类的模型通常是更合适的选择。支持向量机是一种强大的回归和分类模型,它使用核函数将不可分离问题转换为更容易解决的可分离问题。SVM可用于执行线性回归和非线性回归,具体取决于所使用的核函数。开发模型的一种好方法是训练一个线性SVM和一个具有径向基函数核的SVM(一种通用的非线性类型的SVM),以量化从非线性模型中可以获得的增益(如果有的话)。非线性方法可以提供更强大的模型,但代价是不容易解释哪些特征正在影响模型,这是引言中提到的一种折衷。
回归中常用的许多模型也用于分类。训练线性SVM和具有径向基核函数的SVM也是分类任务的良好默认起点。可以尝试的另一种传统方法是k近邻分类。作为最简单的分类方法之一,k近邻分类提供了一个有用的基准性能标记,可以与其他更复杂的模型(如SVM)进行比较。另一类鲁棒的非线性方法是基于集成的模型,如随机森林和XGBoost。这两种方法都是强大的非线性模型,并且有提供了特征重要性估计和需要最小的超参数调整的优势。由于提供了特征重要性值的估计和决策树结构,当要从上面概念上了解哪些特征对预测模型贡献最大时,这些模型是一个很好的选择。
对于分类和回归,许多可用的模型往往具有令人困惑的衍生模型和变体模型。试图先验地预测某一特定方法对某一特定问题的适合程度可能是骗人的,相反,采用经验的、试错的方法来找到最佳模型通常是最严谨的方式。使用现代机器学习套件(如scikit-learn),在这些模型变体之间进行更换通常只需要更改一行代码,因此选择最佳方法的一个好的总体策略是训练和优化各种上述方法,并在最终比较其在测试集上的性能之前,选择在验证集上性能最佳的方法。
使用聚类模型。聚类算法的使用(图3e)在生物学中很普遍。k-means是一种强大的通用聚类方法,与许多其他聚类算法一样,需要将聚类数量设置为超参数。DBSCAN是一种替代方法,它不需要预定义簇的数量,但需要设置其他超参数。还可以在聚类之前执行降维,以提高对具有大量特征的数据集的计算性能。
降维。降维技术用于将具有大量属性(或维度)的数据转换为低维形式,同时尽可能保留数据点之间的相对关系。例如,相似的数据点(例如,两个同源的蛋白质序列)在其低维形式上也应该相似;而不同的数据点,例如,不相关的蛋白质序列,应该保持不同。通常选择二维或三维来实现数据分布的可视化,尽管在机器学习中也会用更多的维度作为输入。这些技术包括数据的线性和非线性变换。生物学中常见的例子包括如图3d所示的主成分分析(PCA)、统一流形逼近与投影(UMAP)和t分布随机邻域嵌入(t-SNE)。使用的技术取决于具体情况:PCA保留了数据点之间的全局关系,并且是可解释的,因为每个主成分都是输入特征的线性组合,这意味着很容易理解哪些特征会导致数据的变化。t-SNE更注重保留数据点之间的局部关系,是一种可以揭示复杂数据集结构的灵活方法。应用包括单细胞转录组学中t-SNE 用于细胞分型和PCA 用于分子发育学轨迹分析。
文章参考:https://www.nature.com/articles/s41580-021-00407-0
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























机器学习
后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集



