DeepSS2GO——基于 CNN 的模型可以根据化学键预测蛋白质的功能
导言
论文地址:https://www.biorxiv.org/content/10.1101/2024.03.30.584129v1
关于预测蛋白质功能的方法
预测蛋白质在人体中的功能对于了解生命过程、预防疾病和****开发新药极为重要。特别是近年来,根据蛋白质组成单元的形状和大小、氨基酸序列(称为一级结构)和蛋白质的三维结构(称为三级****结构),开发出了注释蛋白质功能的模型。
然而,一级结构包含过多信息并显示冗余,这限制了其准确预测未知物种蛋白质功能的能力。此外,三级结构在将立体结构信息纳入学习过程时需要巨大的计算成本,因此难以利用大型数据集进行分析。
因此,本文作者提出了DeepSS2GO模型,该模型整合了氨基酸序列信息(一级结构)、根据蛋白质分子中化学键获得的结构特征(这被称为二级结构)以及蛋白质同源性进行学习。该模型不仅比传统算法显示出更好的预测性能,而且减少了所需的计算量。
蛋白质的一级、二级和三级结构
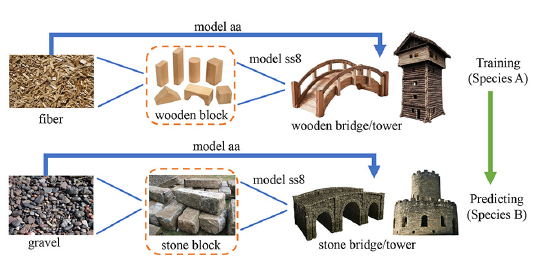
为了更好地了解蛋白质的一级、二级和三级结构,本文将它们与我们在日常生活中看到的构件进行了比较。
在图中,建筑材料、纤维和石块的排列方式相当于一级结构,用这些材料制成的砌块的形状相当于二级结构,而桥梁、塔楼或其他由砌块组成的结构则相当于三级结构。
传统模型 "**模型 aa "**根据一级结构(纤维和砾石)的特征预测三级结构。然而,仅从一级结构(纤维和砾石)的排列来预测成品(桥梁或塔楼)的模式和功能是很困难的。
相比之下,本文提出的模型 "model ss8"可通过二级结构(木块特征)预测三级结构(桥和塔)。换句话说,它声称能够通过有效利用二级结构(木块或石块)进行学习,从而更准确地预测蛋白质的功能。
实验还表明,模型的训练在物种 A 中进行,模型的预测在物种 B 中进行。需要强调的是,在不同物种之间进行训练和预测可以实现高度通用的预测。
利用深度学习预测蛋白质功能
预测蛋白质功能的方法可根据所使用的信息源或算法进行分类。使用的信息来源包括一级结构、三级结构和蛋白质-蛋白质相互作用。基于算法的方法包括不使用深度学习的序列同源性比对和深度学习模型(自然语言处理模型),在实践中通常将两者结合使用。
在本实验中,使用基因本体(Gene Ontology,GO)来表示蛋白质的功能,这是一种用有向图表示分子功能、细胞成分和生物过程的方法。由于蛋白质的功能并不是相互独立的,而是往往有相似的部分,因此采用图形表示它们之间的关系。
在本文的模型中,输出结果是一个分数,用介于 0 和 1 之间的数字表示,表示与每个功能相关的词(在下面的模型结构图中对应于 GO1、GO2 等)是否具有一个功能。
型号详情
模型结构
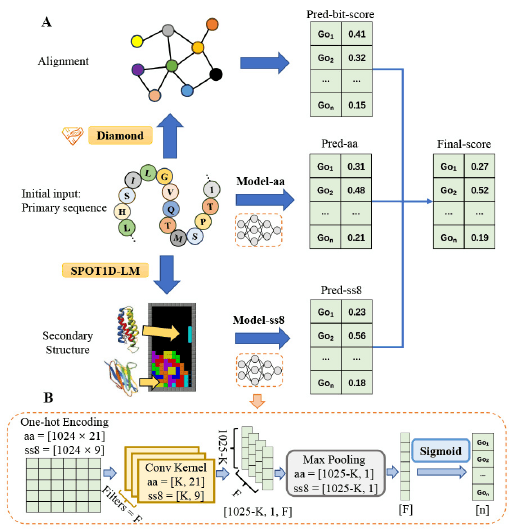
DeepSS2GO模型的整体示意图如上图所示。
在训练过程中,首先要对数据进行预处理,获取预过滤蛋白质的一级结构及其注释。请注意,蛋白质序列和注释是从 SwissProt 和 CAFA3 这两个数据集收集的,并在本实验中用作输入数据。
接下来的任务是使用图中的 "SPOT1D-LM "算法从一级结构预测二级结构。请注意,一级结构由20 个不同的字符组成,二级结构由8 个不同的字符组成。
输入包含 1024 行一级结构和二级结构的序列信息,并以单击矩阵的形式表示,其中一级结构 21 列,二级结构 9 列。
一级结构由 "**模型-aa "**处理,并输出预测得分(图中为 Pred-aa)。二级结构由 "模型-ss8"处理,并输出预测得分(图中为 Pred-ss8)。
此外,"钻石法"工具还用于预测蛋白质一级结构的同源性。通过预测得到的分数(图中的 Pred-bit-score 分数)将被输出。(请注意,Diamond 方法并不使用机器学习模型,而是使用传统生物信息学中的方法)。
然后对通过主序列、次序列和钻石法获得的三种预测分数(即 Pred-ss8、Pred-aa 和 Pred-bit-score)进行整合。整合三种分数时使用了预先指定的参数。
输入数据经过多个具有不同内核大小和滤波器的卷积神经网络。然后通过一个 McSpooling 层进行归一化处理,并通过一个 sigmoid 函数进行激活,使输出保持在 0 到 1 的范围内。请注意,在模型训练过程中引入了提前停止功能,以防止训练过度。
这里,K代表内核的宽度,本实验使用了不同的 K 值。
实验结果
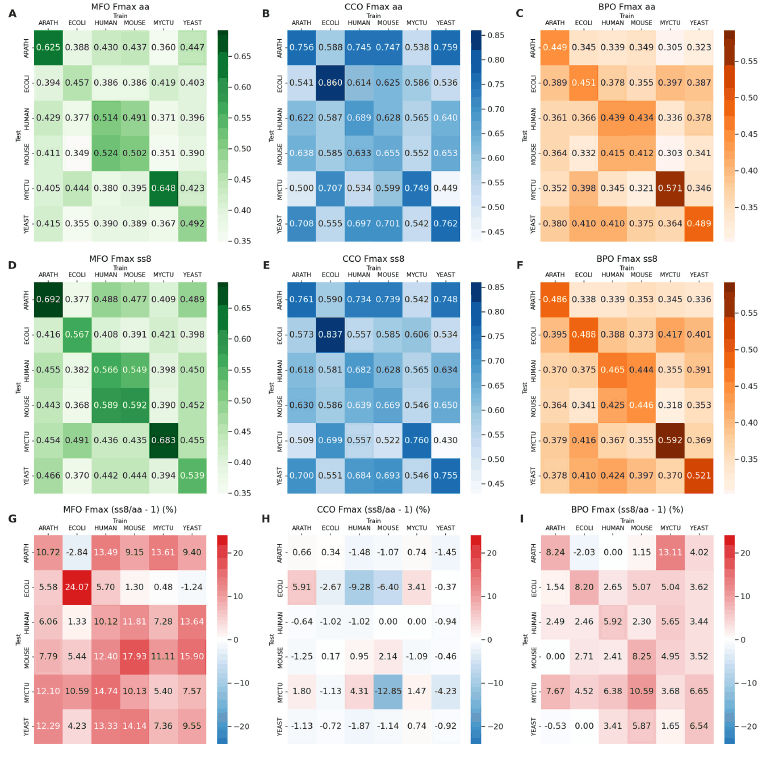
上图以分数图的形式显示了Fmax,即准确度和重现性的谐波平均值的最大值,作为评估模型的指标。 请注意,在本实验中,如前所述,训练数据和测试数据的测试方式是在不同物种间交叉进行的:六行和六列代表不同物种,包括人类;例如,如果纵轴为人类,横轴为鼠标,则表示人类数据用于测试数据,而鼠标数据用于训练数据。数据用于训练数据。
得分图显示,颜色越深,Fmax 分数越高(即模型性能越好)。 图 A 至图 C显示了基于一级结构的模型-aa 的评估结果。图 D 至 F 则显示了基于二级结构的模型-ss8 的评估结果。
具体来说,图 A、D 和 G 基于分子的功能,图 B、E 和 H 基于细胞所含的成分,而图 C、I 和 F 则基于生物学过程。 另一方面,图 G 至图 I显示了基于二级结构的模型(Model-ss8)比基于一级结构的模型(Model-aa)要好得多。
红色越深,表明基于二级结构的模型性能越好。 实验结果表明,与仅使用一级结构信息相比,使用蛋白质二级结构信息可以提高蛋白质功能预测任务的准确性。
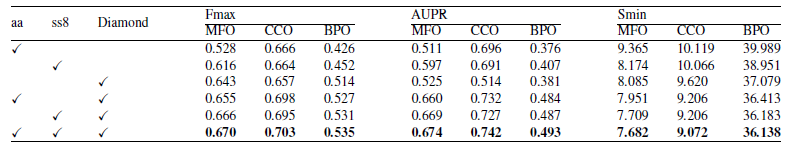
上表评估了上述模型的性能。下一行显示的是该模型的方法论–DeepSS2GO。
除了前面提到的 Fmax 外,使用的评价指标还有AUPR和Smin,前者代表准确度重现性曲线下的面积,后者通过计算真阳性率和假阳性率之间的差值来评估模型区分阳性的能力。
在使用不平衡数据集时,AUPR是一个有用的指标,用来评估模型是否能够准确识别少数阳性病例,因为错误分类阳性病例的惩罚更大。
另一方面,Smin是通过计算真阳性率和假阳性率之间的差值来衡量模型的判别能力。换句话说,这个值越小,模型区分正负结果的能力就越强。
利用 Fmax、AUPR 和 Smin 等指标,可以确认 DeepSS2GO 方法与传统模型相比具有很高的性能。
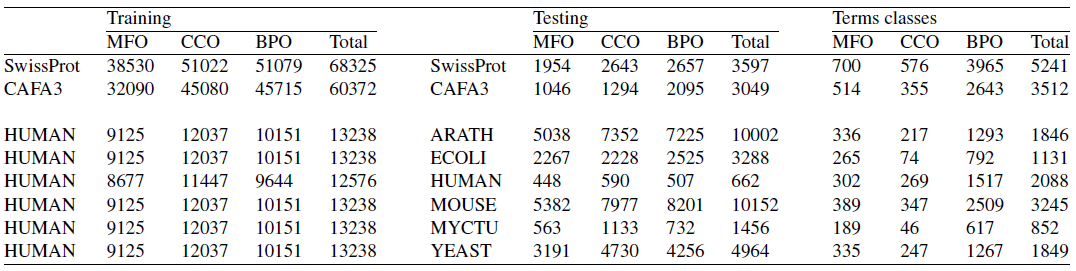
上表显示了仅使用 Model-aa、Model-ss8 和 Diamond 方法中的部分学习模块进行训练时的结果,而当所有三种学习模块都存在时,训练结果最佳。
当使用两种学习模块时,Model-ss8 和 Diamond 的组合往往更高,这表明使用深度学习预测二级结构的模型与传统生物信息学方法 Diamond之间有很好的匹配。
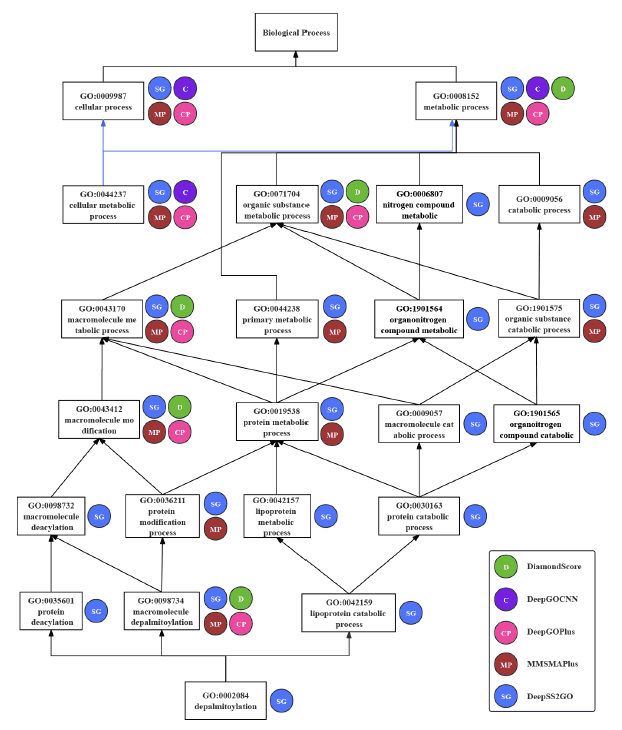
上图显示了DeepSS2GO方法的功能预测结果。每个方框都包含用于描述功能的术语,最上面方框中的术语与最下面方框中的术语有包含(相同或部分包含)关系。
每个节点上显示的彩色圆圈表示不同的预测方法是否能够预测特定功能(即方框旁边的彩色圆圈表示模型能够预测该功能)。
蓝色圆圈表示拟议方法(DeepSS2GO)的功能预测结果,其他彩色圆圈表示现有方法的功能预测结果。如图所示,拟议方法能够预测多种蛋白质的功能,包括低层的各种功能。
总结
研究发现,DeepSS2GO通过减少主序列中的冗余信息并引入一个整合二级结构特征的学习模块,提高了预测蛋白质功能的性能。
作者引入了经典的卷积神经网络来提高该模型二次结构的有效性,但认为可以利用GNN 和****自监督学习来进一步提高功能预测模型的性能。
此外,用于预测一级结构和二级结构的算法认为,氨基酸长度超过 1024 个的大型蛋白质不在预测范围之内。今后,必须引入对更长序列进行二级结构预测的方法。
此外,该模型还可用于预测多肽的功能,从而更广泛地阐明疾病和发现药物靶点。我个人认为,在预测是否存在特定多肽结构的任务中,该模型可能具有潜在的应用价值。