7+WGCNA+机器学习+实验+泛癌分析,多要素干湿结合
今天给同学们分享一篇生信文章“Analysis and Experimental Validation of Rheumatoid Arthritis Innate Immunity Gene CYFIP2 and Pan-Cancer”,这篇文章发表在Front Immunol期刊上,影响因子为7.3。

结果解读:
DEG筛选和数据预处理
数据在箱线图中进行了标准化,不同的颜色代表不同的数据集,行代表样本,列代表样本中的基因表达值(图1A)。图1B展示了批次去除前多个数据集的PCA结果,不同的颜色代表不同的数据集。如图所示,三个数据集分别分开,没有任何交集。图1C展示了批次去除后的PCA结果图。如图所示,三个数据集的交集可以作为后续分析的一批数据。根据P-adjustment <0.05和log2 fold-change (FC) | >0.5的标准,鉴定出了891个差异表达基因(DEGs),其中427个基因上调,464个基因下调。图1D展示了DEGs的火山图以及前50个基因的热图(图1E)。

DEGs功能富集分析
所有DEG在功能上都得到了富集,根据p<0.05,GOCircle图中显示了15个GO关键词。研究结果表明,生物过程(BP)富集主要与阳性细胞-细胞粘附调节、T细胞活化、淋巴细胞分化和细胞-细胞黏附调节有关。富集分子功能(MF)与细胞因子受体结合、细胞因子结合和细胞因子受体活性有关。细胞成分(CC)富集与质膜外侧、膜筏和膜微区有关。在KEGG分析中,造血细胞谱系、人类T细胞白血病病毒1型感染、Th1和Th2细胞分化以及趋化因子信号通路是相关的。
加权基因共表达网络构建
从GEO数据中检索到GSE1919和GSE55457数据集,并选择了15个正常样本和18个RA样本来对样本进行聚类,并通过设置阈值来排除明显异常的样本,如图2A所示。然后,如图2B所示,当R > 0.9且平均连接性较高时,作者将软阈值设置为7。通过使用0.25的聚类高度限制合并强相关的模块(图2C),共鉴定出24个模块进行进一步研究。最终,在聚类树下显示了经过调整和合并的模块(图2D)。接下来,对模块之间的相关性进行了检查,结果显示它们之间没有显著的关联(图2E)。通过模块内的转录相关性分析证明了模块划分的可靠性,结果显示模块之间没有实质性的联系(图2F)。使用ME值和临床特征之间的前额相关性来研究模块与临床症状之间的关联。蓝色模块与正常样本呈正相关(r = 0.79, p = 5e−08),与RA样本呈负相关(r = −0.79, p = 5e−0)。8),而蓝绿色模块与正常情况呈负相关(r = 0.8,p = 3e−08),与RA呈正相关(r = −0.8,p = 3e−08)(图2G)。临床上有意义的模块被确定出来。结果显示,蓝色和蓝绿色模块在对照组MM与GS散点图(图2H)以及RA MM与GS散点图(图2I)中与RA高度相关。进一步研究了这两个模块中的所有基因。
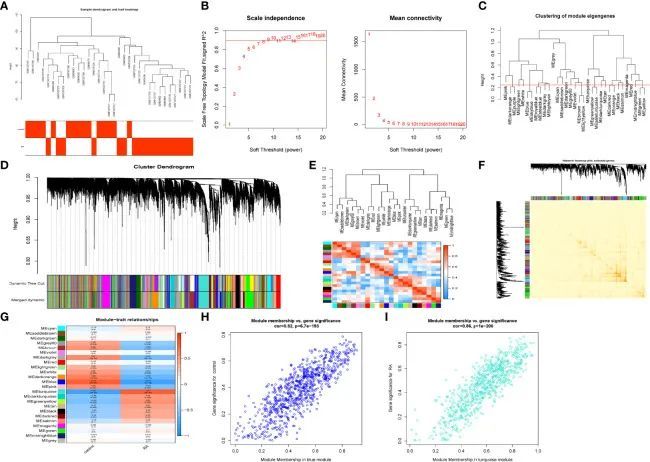
关键模块基因的DEGs和功能分析
在使用维恩图交集关键模块基因和差异表达基因(DEG)基因后,作者发现了490个交集基因(图3A)。作者进行了功能分析,以了解模块中DEG基因的生物学功能。DO分析的结果显示这些DEG与淋巴母细胞白血病、肝炎、生殖细胞癌和造血系统疾病有关(图3B)。GO富集分析显示模块DEG基因具有T细胞激活、细胞间粘附调节、细胞激活的正调节、负向调节细胞激活、细胞外质膜、膜漂浮区、膜微区、细胞因子受体结合、抗原结合和免疫受体活性(图3C)。KEGG分析与细胞因子-细胞因子受体相互作用、趋化因子信号通路和人类免疫缺陷病毒1型感染有关(图3D)。
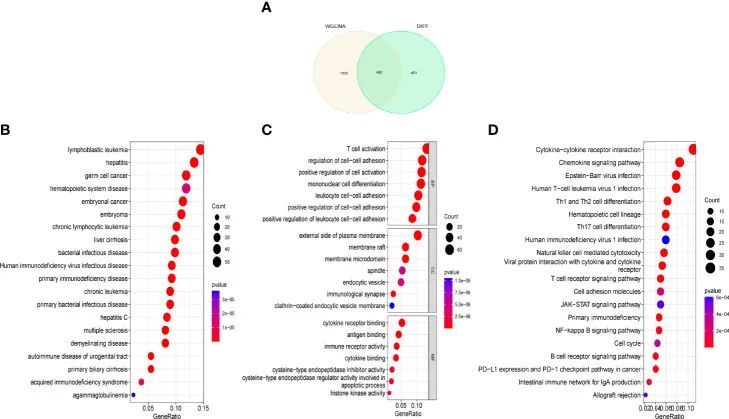
特征基因的选择
作者使用了三种机器算法来识别特征基因:SVM-RFE(图4A,B);LASSO回归分析从统计学上显著的单变量中选择了19个预测基因(图4C);以及RandomForest结合特征选择来确定错误率、分类树数量之间的关系(图4D,E),以及31个具有相对重要性的基因。作者使用Venn图找到了通过上述三种方法的交集交集的四个基因(图4F)。
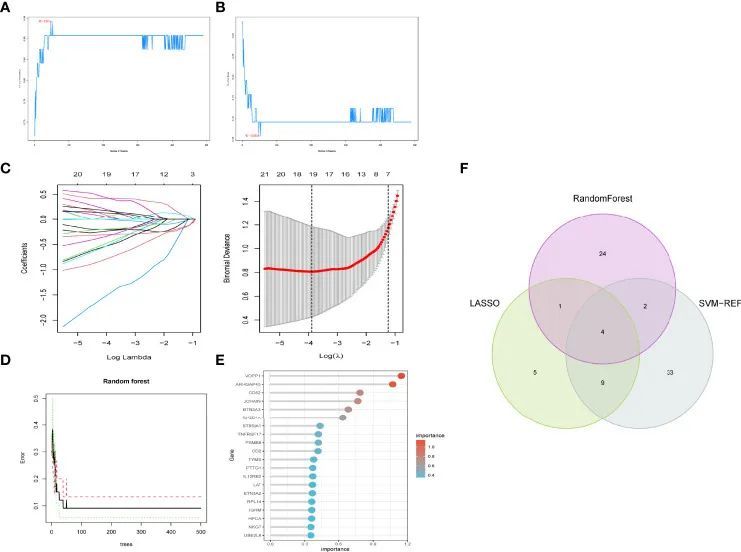
特定基因表达的验证
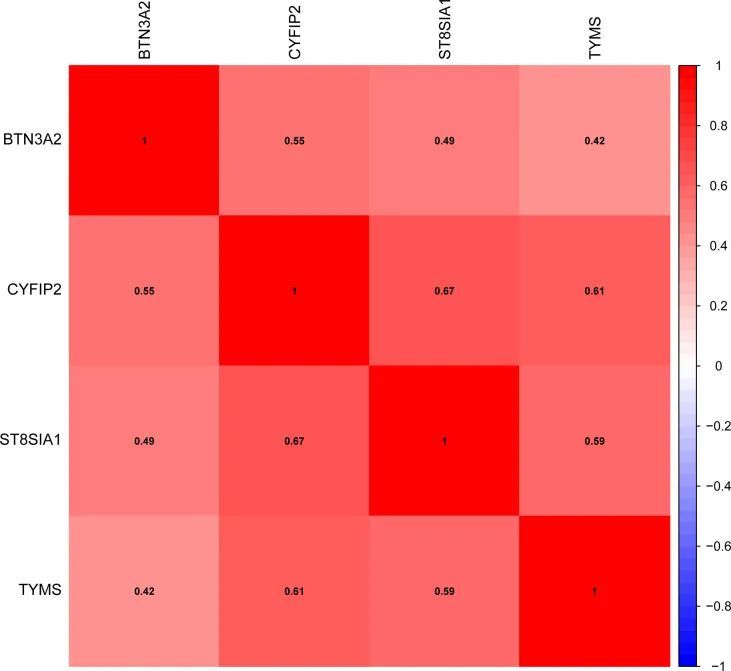
作者使用GSE1919和GSE55447的数据确认了这四个基因在类风湿关节炎中的表达,并发现BTN3A2、CYFIP2、ST8SIA1和TYMS在类风湿关节炎中都显著升高。此外,验证数据集(GSE48780和GSE55235)表明BTN3A2、CYFIP2、ST8SIA1和TYMS在类风湿关节炎中也有显著表达。基因相关性也进行了检查,如图5所示,BTN3A2、ST8SIA1、TYMS和CYFIP2呈正相关,表明这四个基因具有显著的功能相似性。
使用GSEA分析特征基因
为了更好地理解RA中的特征基因的作用,作者使用GSEA将RA组织根据特征基因的中位数表达分为两类。核苷酸代谢、原发性免疫缺陷、嘧啶代谢和视黄醇代谢在高BTN3A2亚组中显著富集,而醛固酮调节的钠重吸收、HIF-1信号通路、氮代谢和肾细胞癌在低BTN3A2亚组中显著富集。可卡因成瘾、甘油脂质造血细胞系谱、免疫网络生产和原发性免疫缺陷在高CYFIP2亚组中显著富集,而移植排斥、肠道IgA产生的免疫网络、烟酸和烟酰胺代谢以及原发性免疫缺陷在低CYFIP2亚组中显著富集。铁死亡、亚油酸代谢、氮造血细胞系谱、肠道免疫网络对IgA产生、原发性免疫缺陷、Th1和Th2细胞分化在高ST8SIA1亚组中显著富集,而铁死亡、亚油酸代谢、氮造血细胞系谱、肠道免疫网络对IgA产生、原发性免疫缺陷、Th1和。高TYMS亚组在免疫缺陷、Th1和Th2细胞分化方面富集,而低TYMS亚组在ABC转运体、昼夜节律、糖酵解/糖异生和近曲小管碳酸氢盐回收方面显著富集。
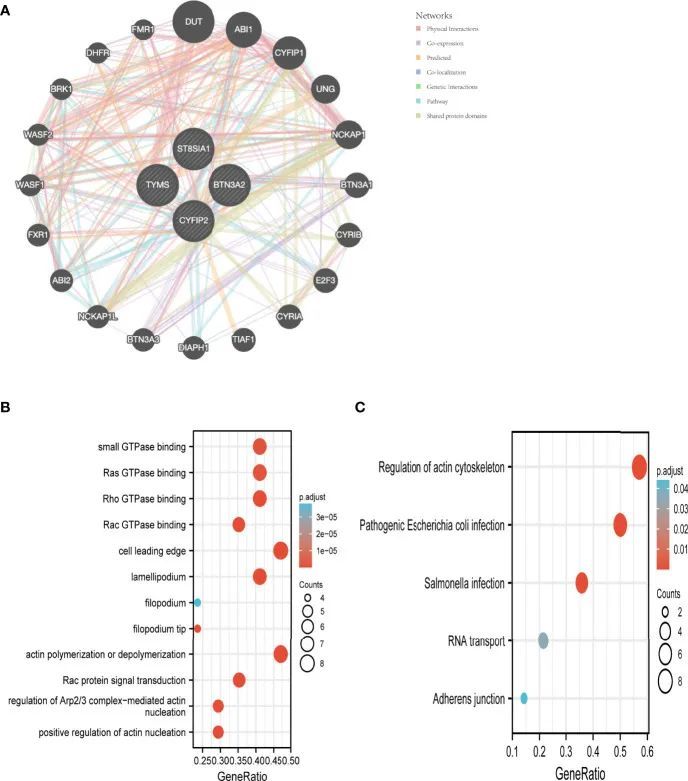
特征基因互作分析
作者使用GeneMANIA数据库为特征基因创建了一个PPI网络(图6A)。为了进一步研究这些特征基因的功能,对20个基因进行了GO/KEGG分析。在这个数据集中,肌动蛋白聚合或解聚、Rac蛋白信号传导以及对Arp2/3复合物介导的肌动蛋白核化的控制是最丰富的生物过程。细胞前缘、薄片状突起和须状突起是最丰富的细胞组分(CC)。此外,Rho GTP酶结合、Ras GTP酶结合、小GTP酶结合和Rac GTP酶结合与富集的分子功能(MF)相关联(图6B)。根据KEGG分析,主要富集的通路包括肌动蛋白细胞骨架的调节、致病性大肠杆菌感染和沙门氏菌感染(图6C)。
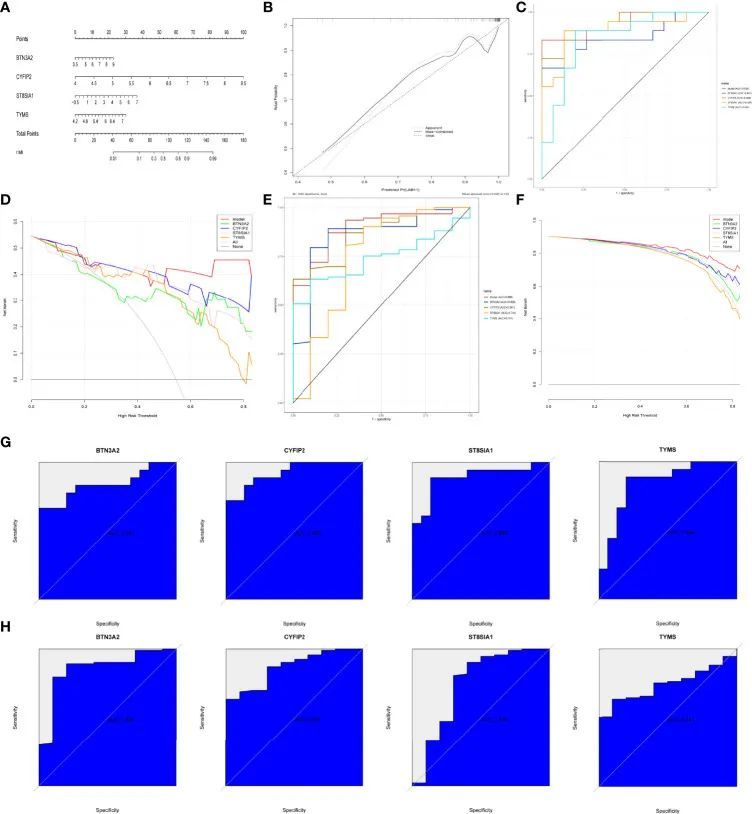
RA诊断柱状线图的建模与测试
作者使用Rms软件包为特征基因(BTN3A2、CYFIP2、ST8SIA1和TYMS)构建了RA诊断柱状线图模型(图7A),并使用校准曲线评估其预测能力。校准曲线显示实际RA风险与预测风险之间的差异非常小,表明柱状线图模型RA非常准确(图7B)。ROC曲线分析也可以确认模型的正确性(图7C)。在决策曲线分析(DCA)中,“柱状线图”曲线高于灰线,而“BTN3A2、CYFIP2、ST8SIA1和TYMS”曲线意味着患者可以在0到1的高风险阈值下从柱状线图模型中获益。柱状线图模型提供了比“BTN3A2、CYFIP2、ST8SIA1和TYMS”曲线更大的临床益处(图7D)。在验证集(GSE48780和GSE55235)中进行的验证也证实了这些发现(图7E、F)。为了进一步验证BTN3A2、CYFIP2、ST8SIA1和TYMS的诊断价值,作者使用受试者工作特征(ROC)分析。BTN3A2(AUC:0.841)、CYFIP2(AUC:0.928)、ST8SIA1(AUC:0.889)和TYMS(AUC:0.发现844)具有类似的AUC值(图7G)。验证数据集(GSE48780和GSE55235)也证实了以下发现:TYMS(AUC:741),BTN3A2(AUC:0.858),CYFIP2(AUC:0.867),ST8SIA1(AUC:0.744)(图7H)。这些发现表明所有主要基因都参与了RA。
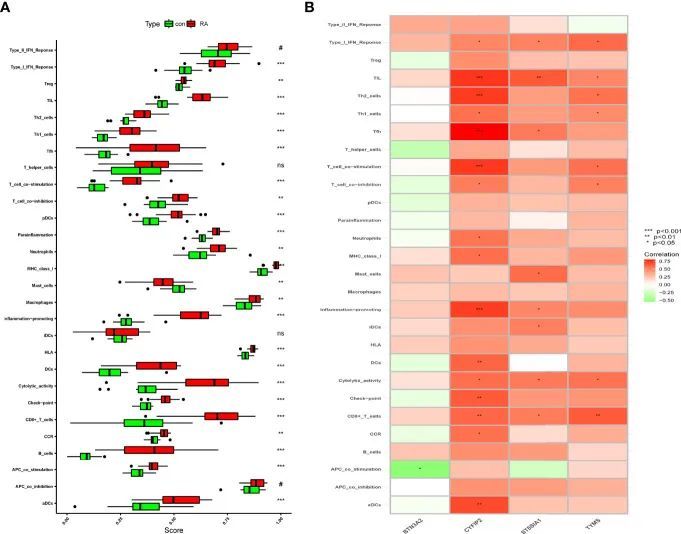
使用ssGSEA分析免疫相关性,研究类风湿关节炎组和健康对照组的免疫浸润情况
进一步使用ssGSEA研究了类风湿关节炎(RA)患者和健康对照组之间的免疫浸润关联。结果显示,在排除非统计学显著性的情况下,肥大细胞和RA中的免疫细胞浸润低于对照组,并且其余RA组中的免疫细胞浸润和免疫相关途径高于对照组(图8A)。作者知道CYFIP2与aDCs、CCR、CD8+ T细胞、检查点、细胞溶解活性、DCs、促炎、MHC I类、中性粒细胞、T细胞共抑制、T细胞共刺激、Tfh、Th1细胞、Th2细胞、TIL和I型干扰素反应相关,并且使用“corrplot”包计算特征基因之间的相关性,结果显示显著正相关(图8B)。BTN3A2与APC共刺激呈负相关。CD8+ T细胞、细胞溶解活性、iDCs、促炎、Tfh、TIL和I型干扰素反应与ST8SIA1均呈强正相关(图8B)。这些特征基因可能在RA进展过程中调节免疫过程。
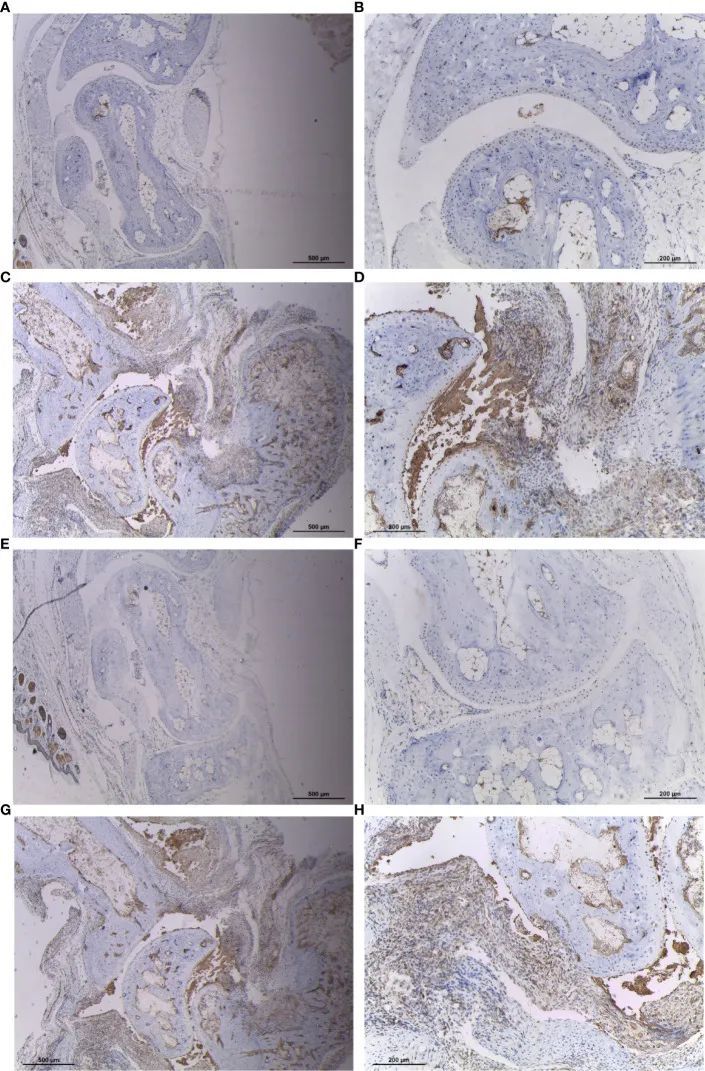
CIA小鼠滑膜组织中CYFIP2和ST8SIA1的表达增加
为了验证CYFIP2和ST8SIA1在类风湿性关节炎滑膜中的表达情况,作者使用免疫组化方法处理小鼠滑膜,并发现CYFIP2和ST8SIA1CIA小鼠在滑膜中高度表达(图9)。
泛癌症CYFIP2表达
免疫基因从InnateDB数据库中提取,并交叉四个标志基因以产生两个交集基因(CYFIP2,ST8SIA1)。在合并ssGSEA结果后,作者将CYFIP2基因提升到下一个分析水平。由于免疫反应不仅在类风湿性关节炎中至关重要,而且在癌症中也很重要,作者使用交集的免疫基因来查看这两种疾病之间是否存在联系。根据TCGA数据(图10A),发现CYFIP2在BRCA、CHOL、HNSC、PRAD、THCA中高表达,在BLCA、BRCA、COAD、ESCA、GBM、KICH、KIRC、KIRP、LUAD、LUSC和PAAD中低表达。作者还从GTEx数据库下载了正常组织数据,并发现CYFIP2在BRCA、CHOL、COAD、DLBC、ESCA、HNSC、OV、PAAD、PCPG、PRAD、READ、SKCM、TGCT、THCA和THYM中强烈表达,而在BLCA、CESC、GBM、KICH、KIRC、KIRP、LGG、LIHC和LUAD中表达较弱(图10B)。正如数据所示,CYFIP2在细胞系中表达(图10C)。
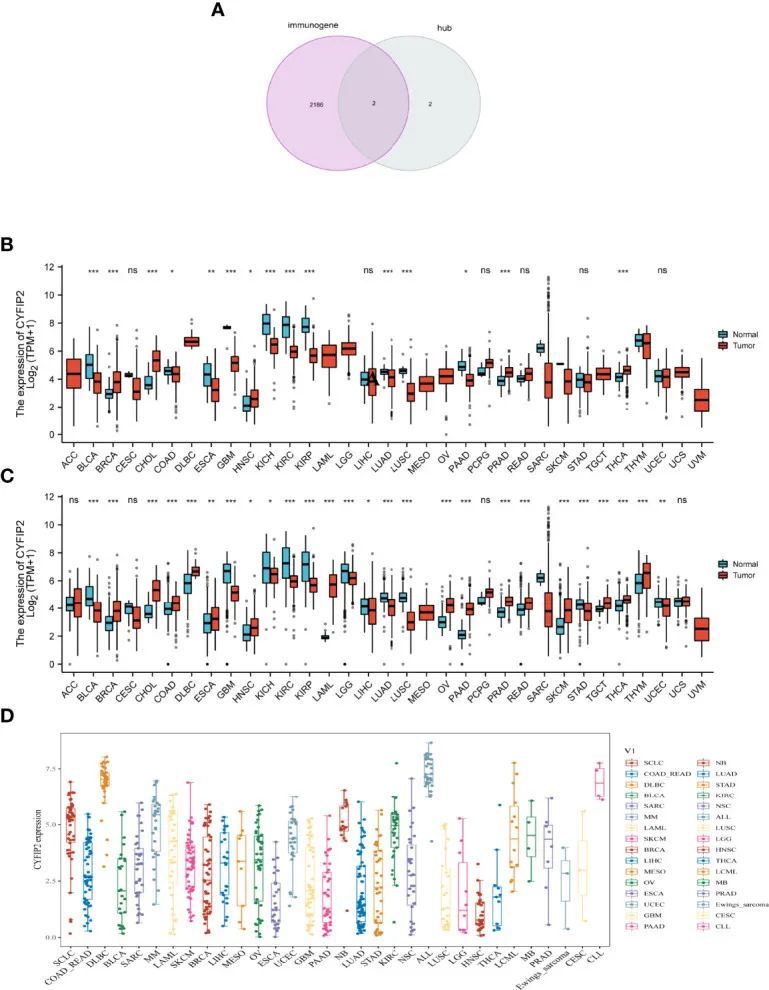
CYFIP2在泛癌中的预后价值
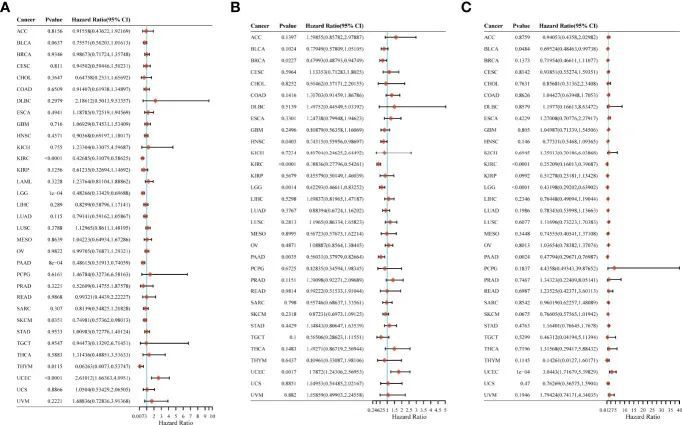
作者研究了CYFIP2表达与全癌症患者预后之间的关系,包括总生存期(OS)、疾病特异性生存期(DSS)和无进展生存期(PFS)。在OS分析中,对33种肿瘤进行的Cox回归显示,CYFIP2表达与6种癌症的OS显著相关:KIRC、LGG、PAAD、SKCM和THYM作为保护因素,而UCEC作为风险因素(图11A)。在PFS研究中,对33种肿瘤进行的Cox回归显示,CYFIP2表达与6种恶性肿瘤的PFS显著相关,BRCA、HNSC、KIRC、LGG和PAAD为保护因素,而UCEC为风险因素(图11B)。在DSS分析中,对33种肿瘤进行的Cox回归显示,CYFIP2表达与5种癌症的DSS显著相关:BLCA、KIRC、LGG和PAAD为保护因素,而UCEC为风险因素(图11C)。
免疫浸润分析
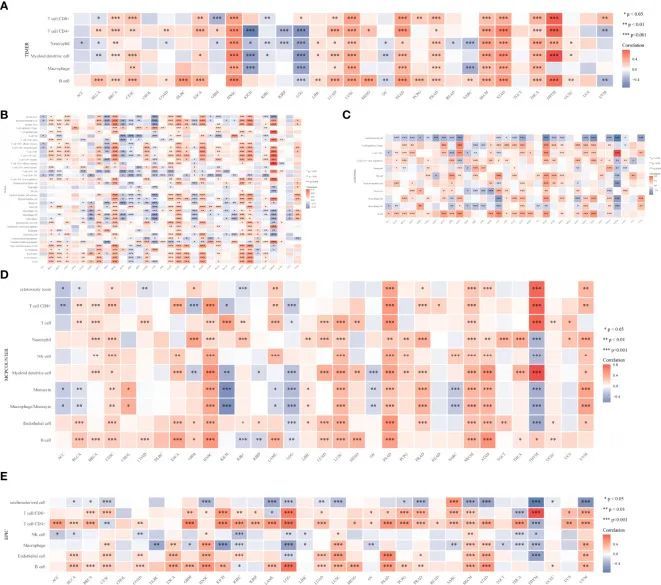
为了更多了解CYFIP2在肿瘤免疫应答中的作用,使用TIMER数据库计算了CYFIP2表达与不同水平的免疫细胞浸润之间的关联。根据研究结果,18个肿瘤中的T细胞CD8+、20个肿瘤中的T细胞CD4+、23个肿瘤中的中性粒细胞、19个肿瘤中的髓样树突状细胞、12个肿瘤中的髓样树突状细胞以及23个恶性肿瘤中的B细胞显示出强烈的关联。HNSC、LUSC、PAAD、SKCM、STAD、THCA和THYM显示出显著的正相关,而KICH和LGG显示出显著的负相关(图12A)。使用xCELL算法(图12B)、QUANTISEQ算法(图12C)、MCPCOUNTER算法(图12D)和EPIC算法(图12E)也证明了CYFIP2水平与浸润的免疫细胞之间的关联。使用ESTIMATE算法计算了基质评分和免疫评分的估计分数,研究结果显示免疫评分与13种癌症相关,而基质评分与16种肿瘤相关。其中,免疫学评分与HNSC(R = 0.64)、LGG(R = -0.59)和STAD(R = 0.5)最为密切相关。HNSC(R = 0.42),LGG(R = −0.45),和UVM(R = 0.48)与基质评分之间有最强的相关性。CYFIP2水平和免疫检查点在多种癌症中显示高度相关,其中UVM大多数呈正相关,而BLCA、BRCA、COAD、HNSC和PRAD大多数呈负相关,这些在UVM中大多数呈负连接。
总结
为了探索免疫浸润与RA以及泛癌症之间关联的特定关键基因,作者进行了全面深入的分析,以分析相关基因和途径。作者发现的2个关键基因(CYFIP2和ST8SIA1)将拓宽我们对分子机制的理解,并为临床治疗带来更多潜在的治疗靶点,这也需要更多的研究来验证和开发。对于进一步的泛癌症分析,CYFIP2被认为是RA和33种肿瘤中最潜在的靶点,这可能为人类免疫相关疾病甚至癌症的治疗带来希望。